End-to-end examples (MToolBox)
Prerequisites Installation, reference bundles, and all dependencies must be completed beforehand.
Architecture MToolBox supports x86_64 only. ARM-based systems, including Apple Silicon (M1/M2/M3/M4/M5), are not supported.
- MIT single-sample run
- MIT cohort run
1. Before running mtDNA calling you must have a BAM file from WES/WGS
Does it matter if I ran WES/WGS with GATK 3.5 or GATK 4.6? No. CBIcall will detect and use the
bamfiles produced by either version.
Just make sure thatbamfiles are available — FASTQ input is not supported.
CBIcall expects a BAM file from a previous WES/WGS run:
CNAG999_exome
└── CNAG99901P_ex <--- ID taken from here
└── *cbicall_bash_gatk-*_w?s_single_* <- The script expects that you have a BAM file inside this directory
Note on nomenclature Please see this page.
2. Create a parameters file
Create a YAML file, e.g. mit_single.yaml.
Important Please make sure you use the same value for the key
samplethat you used for WES/WGS.
Example:
mode: single
pipeline: mit
workflow_backend: bash
input_dir: CNAG999_exome/CNAG99901P_ex
See Configuration Reference for all YAML keys and supported combinations.
3. Run CBIcall
cbicall run -p mit_single.yaml -t 4
-pselects the YAML parameters file-tsets the number of threads
4. Inspect outputs
After completion, you will find:
CNAG999_exome/CNAG99901P_ex/cbicall_bash_gatk-3.5_mit_single_rsrs_*/
01_mtoolbox/
02_browser/
- Final mtDNA report:
01_mtoolbox/mit_prioritized_variants.txt - mtDNA VCF:
01_mtoolbox/VCF_file.vcf - Browser report:
02_browser/<run-id>.html
See Outputs for the full file reference.
5. Visualize variants in the browser
Please see:
02_browser/README.txt
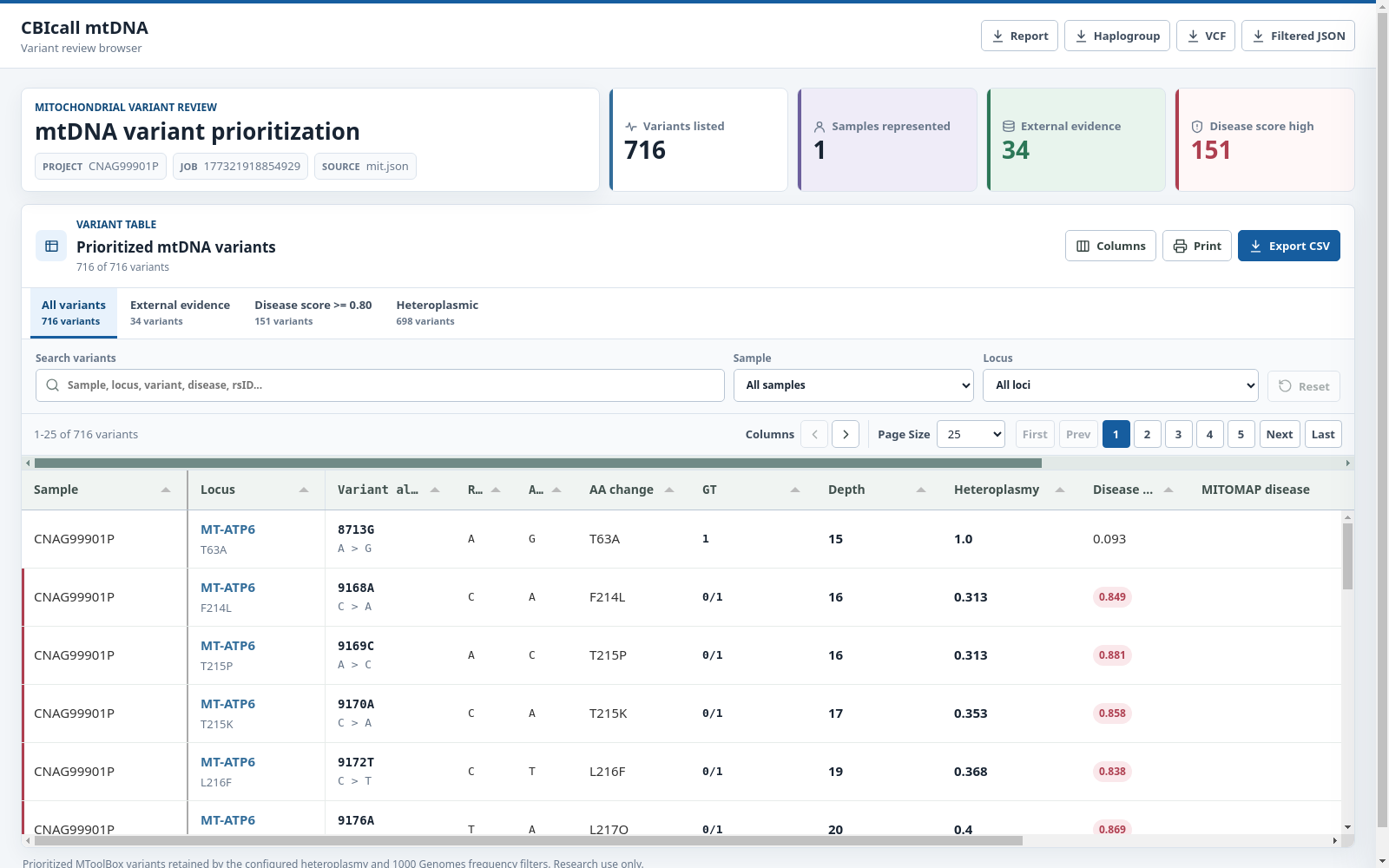
The CBIcall mtDNA variation browser is a standalone HTML report. CBIcall derives
the displayed rows from 01_mtoolbox/mit.filtered.json and embeds them with the
Tabulator table assets at generation time.
Therefore, 02_browser/<run-id>.html opens directly without a local web server,
internet connection, or external static assets.
See snapshot
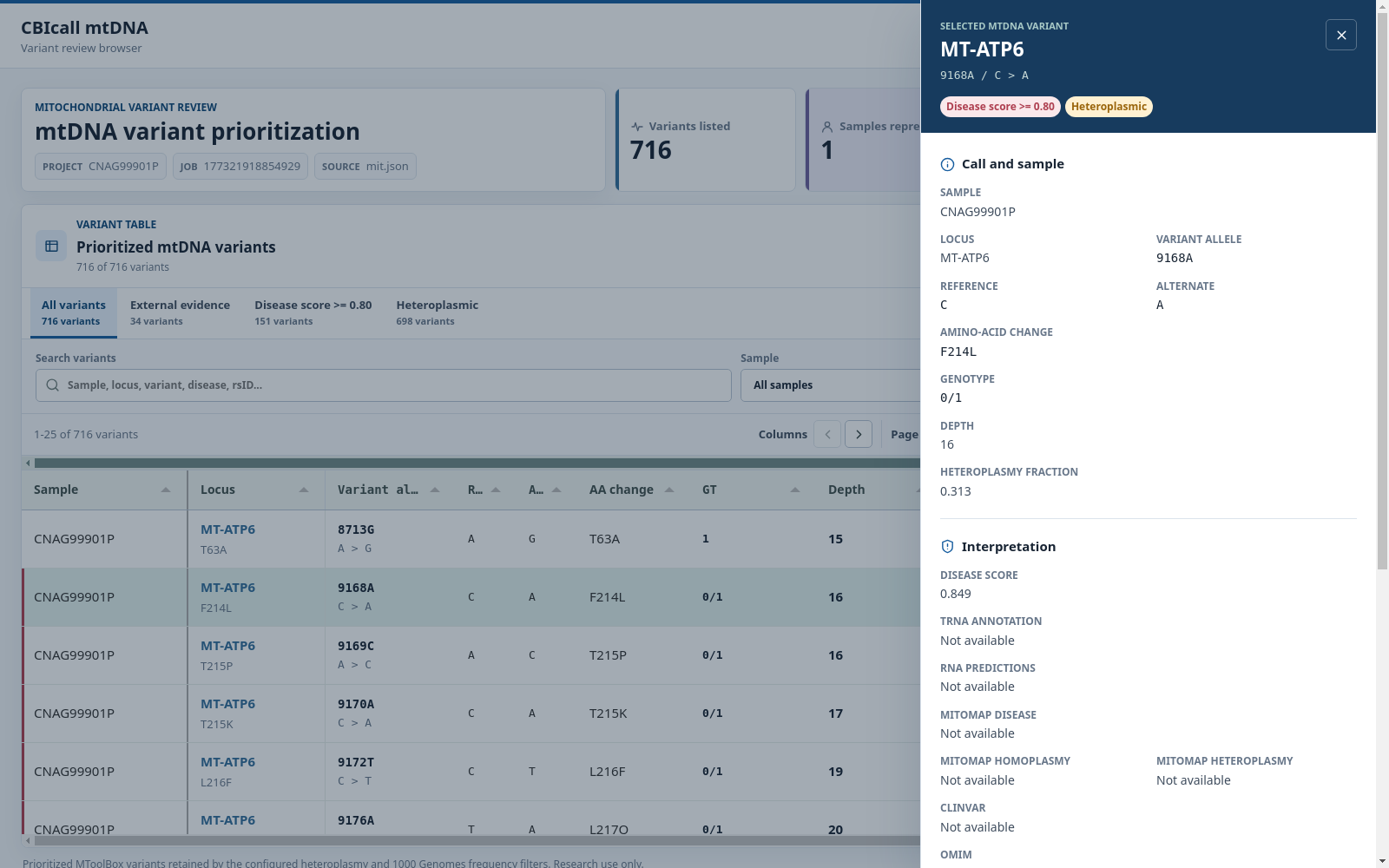
Variant details
Browser actions
The report provides direct buttons for:
- Report:
01_mtoolbox/mit_prioritized_variants.txt, including annotations plus appendedGT,DP, and heteroplasmy values. - Haplogroup:
01_mtoolbox/mt_classification_best_results.csv, including the predicted haplogroup for each sample. - VCF:
01_mtoolbox/VCF_file.vcf, containing the mtDNA variants in VCF format. - Filtered JSON:
01_mtoolbox/mit.filtered.json, containing the records retained by the browser's HF and population-frequency filters.
Quick-filter tabs isolate all variants, external evidence, high disease scores, or heteroplasmic calls. Users can also search across annotations, filter by sample or locus, choose visible columns, sort and paginate records, move horizontally through wide tables, inspect a variant in a detail drawer, print the table, or export the current view as CSV. Disease, evidence, and heteroplasmy indicators retain the same color coding in the table and detail drawer.
HTML table:
The CBIcall mtDNA variation browser displays a browsable table consisting of the most relevant fields relative to the variant annotation:
- Sample: The full name of each sample.
- Locus: The location on the mitochondrial chromosome.
- Variant allele: The position in the mitochondrial chromosome + the alternative allele format.
- Ref: The reference allele (mitochondrial reference genome: RSRS).
- Alt: The alternative allele(s).
- AA change: The amino acid change if the variant falls in a coding region.
- GT: Genotype. 0:Ref, ≥1:Alt(s).
- Depth: The number of times this position is covered by reads.
- Heteroplasmy: The heteroplasmic fraction. Note that the confidence interval can be retrieved from the downloadable VCF file.
- Other: For other fields please consult MToolBox's manual.
Filtered variants The table shows variants retained by the report filters. Variants were excluded if:
- HF ≤ 0.30 (maximum HF observed in any sample)
- 1000 Genomes frequency ≥ 0.01
- Not present in the input VCF
By default, variants with missing HF values (NA,N/A,.) are excluded.
Use the --keep-missing-hf option to retain them.
For advanced parameters, multi-sample analyses, mtDNA workflows and troubleshooting, see the Usage and FAQ sections.
1. Before running mtDNA calling you must have BAM files from WES/WGS
Does it matter if I ran WES/WGS with GATK 3.5 or GATK 4.6? No. CBIcall will detect and use the
bamfiles produced by either version.
Just make sure thatbamfiles are available — FASTQ input is not supported.
CBIcall expects BAM files from previous WES/WGS runs:
CNAG999_exome
└── CNAG99901P_ex <--- ID taken from here
└── *cbicall_bash_gatk-*_w?s_single_* <- The script expects that you have a BAM file inside this directory
CNAG99902M_ex <--- ID taken from here
└── *cbicall_bash_gatk-*_w?s_single_* <- The script expects that you have a BAM file inside this directory
Note on nomenclature Please see this page.
2. Create a parameters file
Create a YAML file, e.g. mit_cohort.yaml:
mode: cohort
pipeline: mit
workflow_backend: bash
software_stack: gatk-3.5
input_dir: CNAG999_exome
See Configuration Reference for all YAML keys and supported combinations.
3. Run CBIcall
cbicall run -p mit_cohort.yaml -t 4
-pselects the YAML parameters file-tsets the number of threads
4. Inspect outputs
After completion, you will find:
CNAG999_exome/cbicall_bash_gatk-3.5_mit_cohort_rsrs_*
01_mtoolbox/
02_browser/
- Final joint mtDNA report:
01_mtoolbox/mit_prioritized_variants.txt - Joint mtDNA VCF:
01_mtoolbox/VCF_file.vcf - Browser report:
02_browser/<run-id>.html
See Outputs for the full file reference.
5. Visualize variants in the browser
Please see:
02_browser/README.txt
The CBIcall mtDNA variation browser is a standalone HTML report. CBIcall derives
the displayed rows from 01_mtoolbox/mit.filtered.json and embeds them with the
Tabulator table assets at generation time, so
02_browser/<run-id>.html opens directly without a local web server, internet
connection, or external static assets. The cohort report follows the same
format as the single-sample report, with sample-level filtering and fields.
Browser actions
The report provides direct buttons for:
- Report:
01_mtoolbox/mit_prioritized_variants.txt, including annotations plus appended per-sampleGT,DP, and heteroplasmy values. - Haplogroup:
01_mtoolbox/mt_classification_best_results.csv, including the predicted haplogroup for each sample. - VCF:
01_mtoolbox/VCF_file.vcf, containing the mtDNA variants in VCF format. - Filtered JSON:
01_mtoolbox/mit.filtered.json, containing the records retained by the browser's HF and population-frequency filters.
Quick-filter tabs isolate all variants, external evidence, high disease scores, or heteroplasmic calls. Users can also search across annotations, filter by sample or locus, choose visible columns, sort and paginate records, move horizontally through wide tables, inspect a variant in a detail drawer, print the table, or export the current view as CSV. Disease, evidence, and heteroplasmy indicators retain the same color coding in the table and detail drawer.
HTML table:
The CBIcall mtDNA variation browser displays a browsable table consisting of the most relevant fields relative to the variant annotation:
- Sample: The full name of each sample.
- Locus: The location on the mitochondrial chromosome.
- Variant allele: The position in the mitochondrial chromosome + the alternative allele format.
- Ref: The reference allele (mitochondrial reference genome: RSRS).
- Alt: The alternative allele(s).
- AA change: The amino acid change if the variant falls in a coding region.
- GT: Genotype. 0:Ref, ≥1:Alt(s).
- Depth: The number of times this position is covered by reads.
- Heteroplasmy: The heteroplasmic fraction. Note that the confidence interval can be retrieved from the downloadable VCF file.
- Other: For other fields please consult MToolBox's manual.
Filtered variants The table shows variants retained by the report filters. Variants were excluded if:
- HF ≤ 0.30 (maximum HF observed in any sample)
- 1000 Genomes frequency ≥ 0.01
- Not present in the input VCF
By default, variants with missing HF values (NA,N/A,.) are excluded.
Use the --keep-missing-hf option to retain them.
Genetic data interpretation disclaimer: review the project disclaimer before clinical or diagnostic interpretation.