Outputs
CBIcall writes each run into a run directory named from the workflow choices:
cbicall_<backend>_<software-stack>_<pipeline>_<mode>_<genome>_<run-id>/
The exact files depend on the selected pipeline and mode. The tables below are derived from the workflow output definitions and the checked-in example runs.
Native CBIcall pipelines (wes, wgs, and mit) create the run directory
under the discovered sample/input directory. External nf-core workflows are
different: because their inputs are supplied through nfcore_parameters and may
point anywhere, CBIcall creates the run directory in the directory where
cbicall run is launched.
- WES/WGS: use the final QC VCF in
02_varcall/. - WES/WGS single-sample runs: keep the gVCF if you plan cohort joint genotyping.
- mtDNA: use
01_mtoolbox/mit_prioritized_variants.txtand the browser report in02_browser/.
Common Run Files
| File | Meaning |
|---|---|
log.json | Structured record of CLI arguments, resolved configuration, selected runtime profile, compact resources.bundle provenance, and runtime parameters. |
cbicall-execution-contract.json | Backend-ready execution plan created after CBIcall validates and resolves the parameters YAML. It records the command, CBIcall-controlled environment overrides, backend/provider identity, and generated backend launch files. |
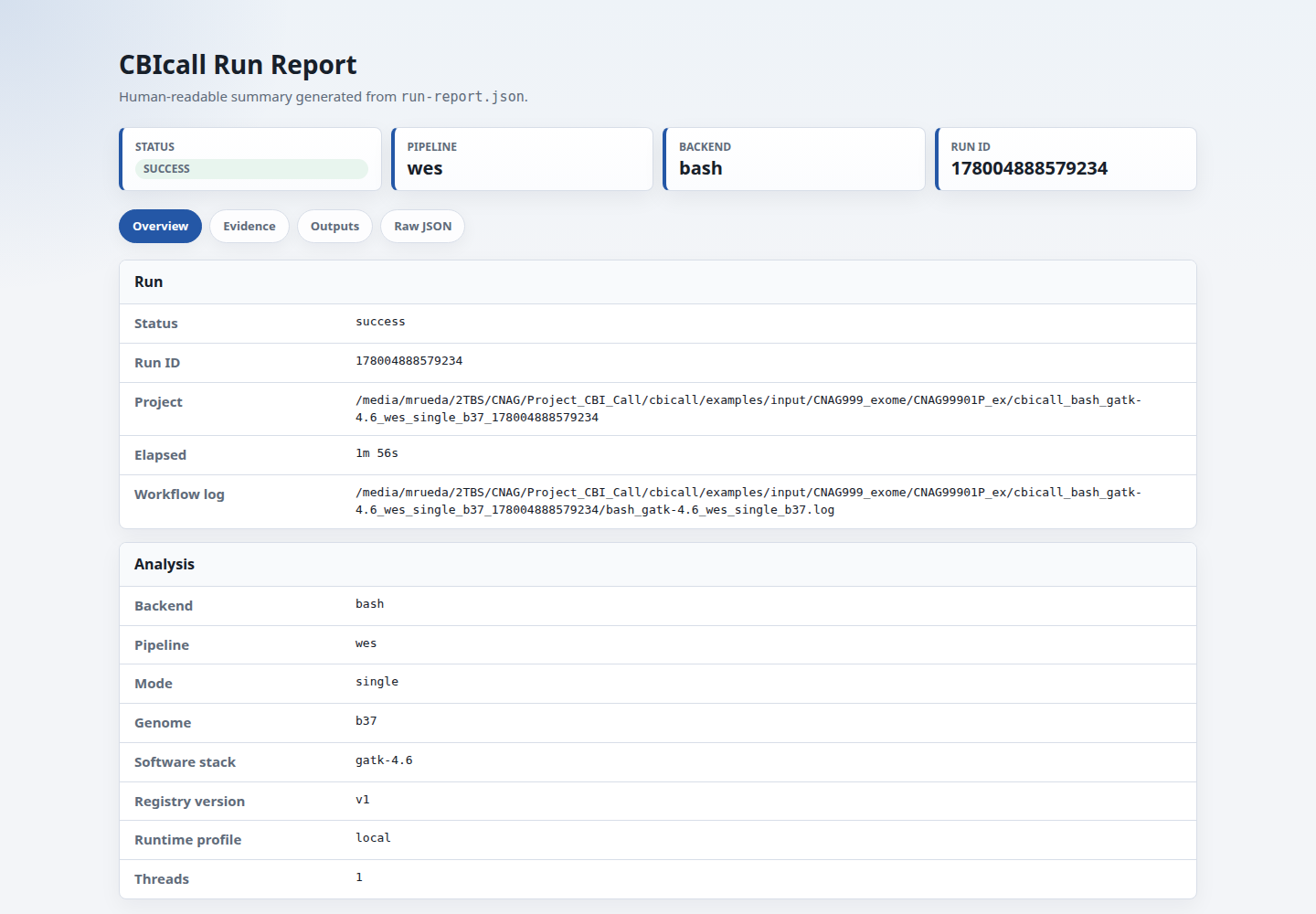
run-report.json | Compact audit report with CBIcall version, hostname, host thread count, Python version, Java version, workflow backend version, status, elapsed time, workflow file fingerprints, execution-contract fingerprint, resource key/version/fingerprint, output file inventory fingerprint, output fingerprints when available, and workflow log path. Runs that fail after execution starts retain the same report with status: failed, error details, and any partial output evidence. |
run-report.html | Human-readable tabbed rendering of run-report.json for browsing a successful or failed run without reading JSON directly. It separates overview, evidence, outputs, and raw JSON views; links the main run evidence; and shows software-version evidence when available. Generate it from an existing run with cbicall report RUN_DIR --html. |
cbicall_mqc/ | Optional MultiQC custom-content directory generated with cbicall run --multiqc, cbicall report RUN_DIR --multiqc, or cbicall compare-runs ... --multiqc. It lets standard MultiQC reports include compact CBIcall run/QC summaries, pairwise comparison tables, and audit-similarity heatmaps without installing a CBIcall MultiQC plugin. |
<backend>_<software-stack>_<pipeline>_<mode>_<genome>.log | Main workflow log for the selected backend. |
logs/*.log | Per-rule or per-step logs for Snakemake/GATK 4.6 workflows. |
Preflight errors that occur before CBIcall creates a run directory, such as an invalid YAML key or an unavailable resource bundle, do not produce a run report.
Use config.resources.bundle.fingerprint inside log.json to check whether two runs used the same declared external dependency set.
Use workflow.fingerprint inside run-report.json to check whether two runs used the same resolved workflow file contents. If the fingerprint differs, inspect workflow.files to see which entrypoint, helper, Snakefile, or config file changed. The matching run-report.html file presents the same core audit fields in a browser-friendly view.
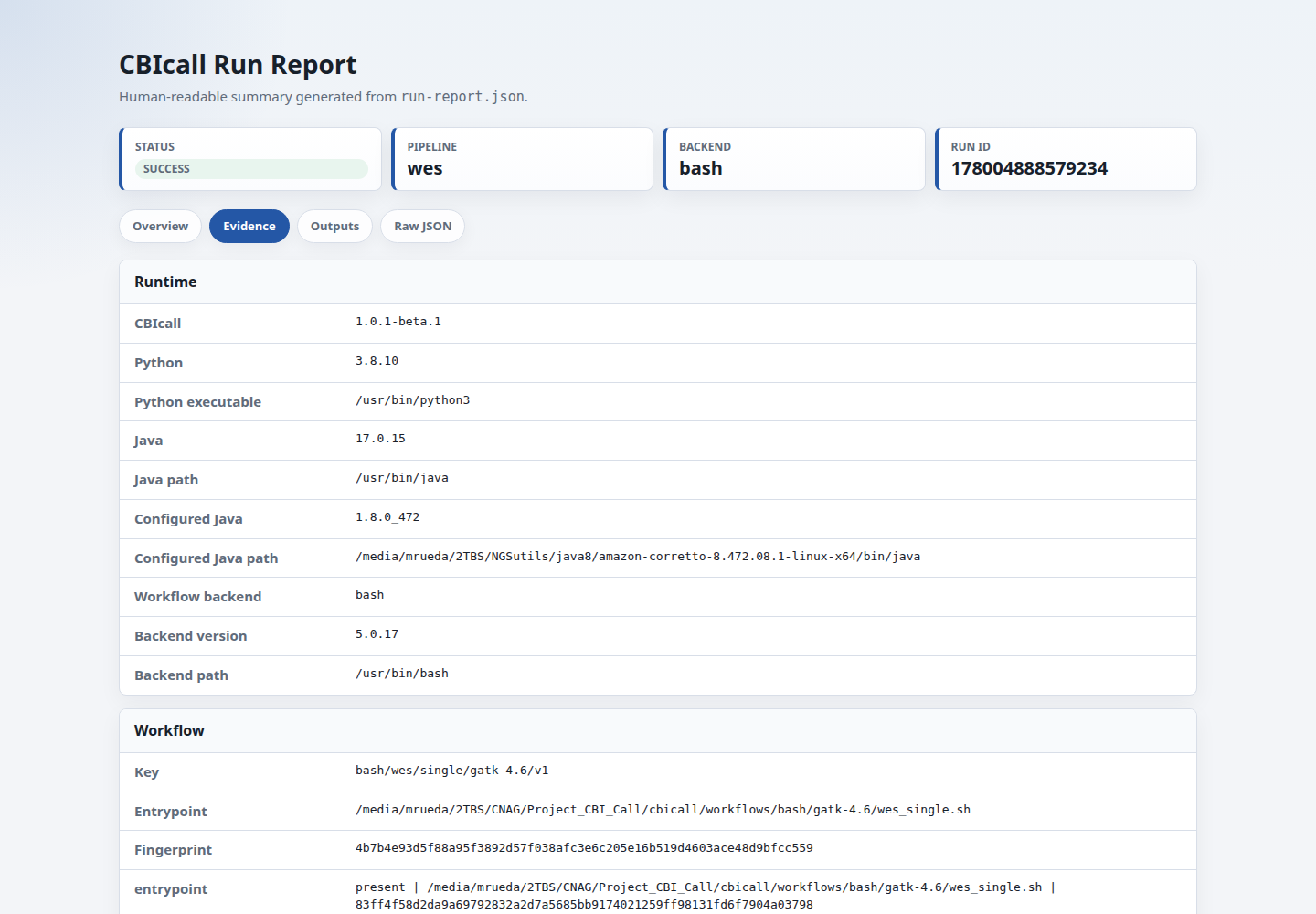
Use runtime.java and runtime.configured_java to audit the Java visible on PATH and the native workflow Java configured through env.sh or shared backend config when available.
Use run.hostname and run.host_threads as runtime context. These fields help
explain where a run happened, but they are not strict reproducibility criteria:
valid cross-machine runs can use different hostnames while producing the same
workflow, resource, execution-contract, and final-output fingerprints.
Use execution_contract.fingerprint to check whether two runs used the same normalized backend-ready execution plan. The raw contract keeps paths and run IDs for audit, while the normalized fingerprint replaces the run directory and run ID so repeated runs can still compare cleanly.
cbicall-execution-contract.json records the normalization placeholders used for fingerprinting. Values such as the concrete run directory and run ID are replaced with {PROJECT_DIR} and {RUN_ID} before hashing, so equivalent repeated launches do not differ only because they were written to different directories.
Use execution_trace to audit task count and peak RAM when the backend emits a trace. For nf-core/Nextflow runs, CBIcall parses pipeline_info/execution_trace_*.txt and records maximum peak RSS and VMEM. Native Bash runs do not have RAM summaries unless the workflow is instrumented to write them.
Use software_versions.sha256 to audit the tool-version table when available. Native workflows use declared tool versions from the selected resource catalog entry. External nf-core workflows use the software-version YAML generated by the nf-core pipeline.
MultiQC Custom Content
CBIcall can write a MultiQC custom-content directory from an existing run report:
cbicall report completed_run/ --multiqc
multiqc completed_run/
The generated cbicall_mqc/ directory contains several small *_mqc.yaml files.
MultiQC renders these as compact CBIcall sections: numeric run statistics,
workflow/resource identity, final-output fingerprints, and native sample QC when
03_stats/*.coverage.txt or 03_stats/*.sex.txt files are present. The full
CBIcall audit remains in run-report.json and run-report.html; MultiQC is a
companion summary for projects that already collect QC with MultiQC. No
CBIcall-specific MultiQC plugin is required. Source installs include multiqc
from requirements.txt so users can render the report directly.
During a new run, use:
cbicall run -p parameters.yaml -t 4 --multiqc
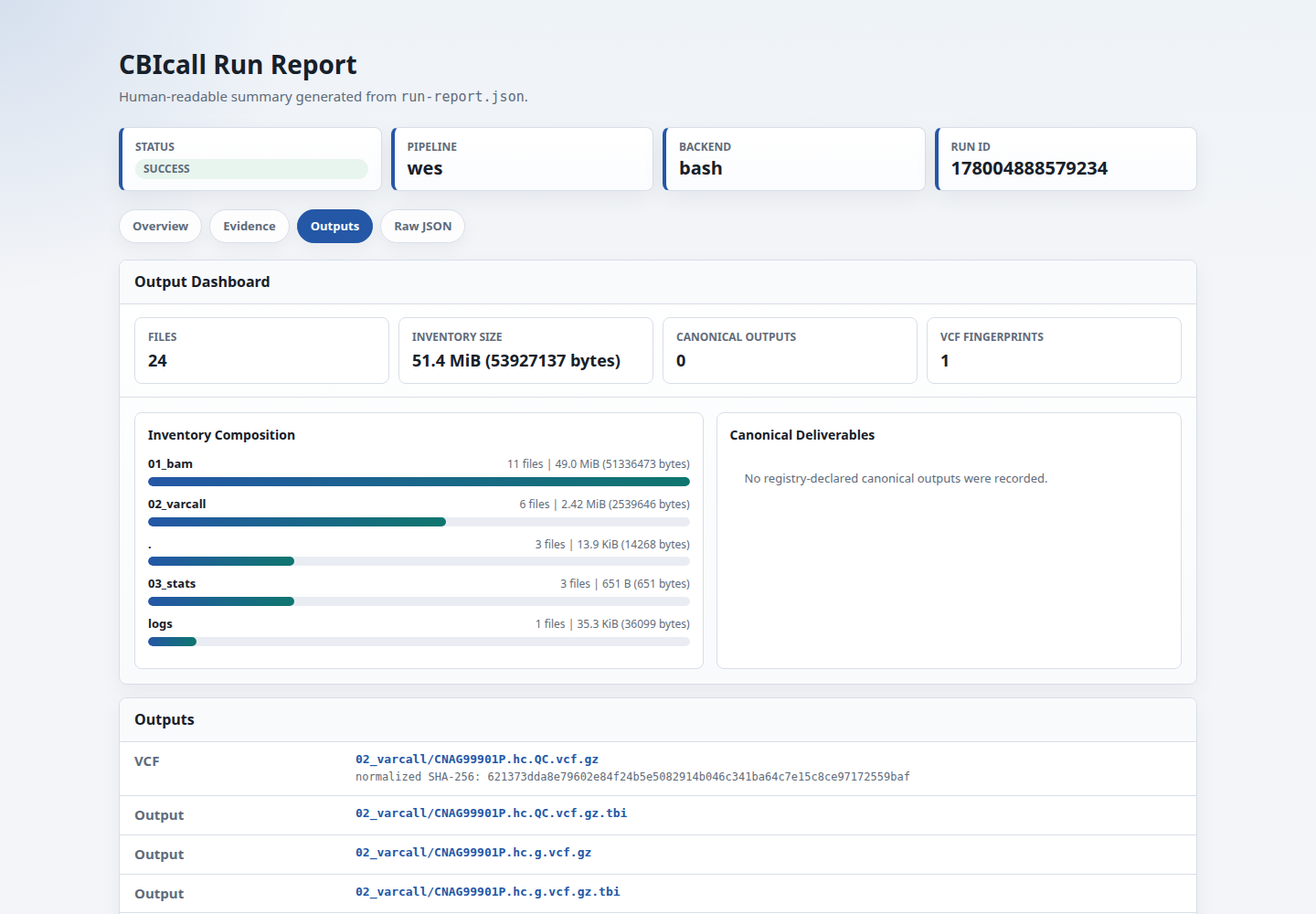
Use outputs.file_inventory.sha256 to check whether two run directories contain the same relative file layout. This is a manifest hash of file paths, not a content hash. outputs.file_inventory.total_bytes records the total size of files included in that inventory; the HTML report renders this in human-readable units and shows the largest files separately so large runs remain readable. WES/WGS single-sample runs also include parsed VCF hash reports under outputs.vcf_hash_reports when 03_stats/*.vcf.sha256.txt is present.
Two runs can be compared directly:
cbicall compare-runs run_a/ run_b/ run_c/ --alias local cloud hpc --output compare-report.txt
The text report is the canonical audit artifact. CBIcall also writes
compare-report.html by default for browsing, including field-level matrices
and combined pairwise audit matrices with derived categories plus report-level
similarity scores. See Run Comparison for details and example
screenshots.
External nf-core Workflows
For workflow_provider: nf-core, CBIcall keeps the upstream nf-core output
layout intact:
| File or directory | Meaning |
|---|---|
cbicall_external_nextflow.params.yaml | Params file generated by CBIcall and passed to nextflow run; its hash is also recorded in the execution contract. |
cbicall_external_nextflow.config | Nextflow config generated by CBIcall to cap process CPU requests from -t/--threads and configure optional container cache paths; its hash is also recorded in the execution contract. |
<pipeline>/ | Upstream nf-core output directory, for example demo/ or sarek/. |
work/ | Nextflow work directory, excluded from the compact run file-inventory hash. |
nf-core_<pipeline>_<mode>.log | Main Nextflow launcher log for the external nf-core workflow. |
run-report.json records the nf-core source, pinned release, nf-core profile,
generated params/config-file hashes, workflow output directory, pointers to
pipeline_info/MultiQC reports, the nf-core software-version YAML, and a
summary of task count and peak RAM from the Nextflow execution trace when
available.
The generated params file also records max_cpus from the CBIcall -t/--threads
value. nf-core parameters such as max_memory can be passed through
nfcore_parameters. The generated Nextflow config applies the CPU value, and
max_memory when present, through process.resourceLimits.
When nfcore_singularity_cache_dir is set, CBIcall writes a user/project-owned
Singularity and Apptainer cache/library path to the generated Nextflow config.
Environment variables such as NXF_SINGULARITY_CACHEDIR belong in the shell or
SLURM bootstrap, not in CBIcall's Python runner.
On ARM64 hosts using the Docker profile, the generated config also pins Docker
to linux/amd64 because many nf-core containers are published primarily for
AMD64.
For registered external workflows, the workflow registry can declare canonical
outputs. The Sarek entry declares the HaplotypeCaller VCF pattern under
sarek/variant_calling/haplotypecaller/. When a matching VCF exists, CBIcall
records it under outputs.canonical_outputs and adds VCF fingerprints to
outputs.vcf_hash_reports for compare-runs.
WES/WGS Single-Sample
Applies to pipeline: wes or pipeline: wgs with mode: single.
Recommended Files
| File | Use |
|---|---|
02_varcall/<id>.hc.QC.vcf.gz | Final filtered single-sample VCF. This is the primary workflow VCF for downstream tools or review. |
02_varcall/<id>.hc.QC.vcf.gz.tbi | Tabix index for the final VCF. |
02_varcall/<id>.hc.g.vcf.gz | Per-sample gVCF. Use this as input for cohort joint genotyping. |
02_varcall/<id>.hc.g.vcf.gz.tbi | Tabix index for the gVCF. |
03_stats/<id>.coverage.txt | Coverage summary with a region-first tabular schema. |
03_stats/<id>.sex.txt | Sex inference result from the final VCF. |
03_stats/<id>.vcf.sha256.txt | Per-VCF SHA-256 report with raw-file, sample count/order, call-level, and strict-record VCF fingerprints. |
Coverage files use one tabular row per sample:
region sampleID mode total_reads mean_cov ten_pct nondup_pct ins_size in_pct out_pct
1 CNAG99901P WES 244 0.0 0.0 0.0 161.5 0.0 100.0
region records the contig used for the lightweight coverage summary. It defaults to chr1 and can be changed with qc_coverage_region in the parameters YAML; this does not change variant-calling intervals. For WES runs, in_pct and out_pct describe reads inside or outside the target definition used by the selected workflow implementation.
Sex files are a lightweight QC signal, not a definitive biological or clinical sex determination method. The helper reports autosomal, X, and Y variant-record depths, the X/autosome ratio, the X-Y depth difference, the threshold, and the decision rule used for the final sex call. It does not require BCFtools; bcftools +guess-ploidy can be used separately as an independent manual cross-check when available.
Intermediate files
| File | Meaning |
|---|---|
01_bam/<fastq-prefix>.rg.bam | Lane-level BAM after alignment and read-group assignment. |
01_bam/<id>.rg.merged.bam | BAM after merging lanes for the sample. |
01_bam/<id>.rg.merged.dedup.bam | Duplicate-marked BAM. |
01_bam/<id>.rg.merged.dedup.metrics.txt | Duplicate-marking metrics. |
01_bam/<id>.rg.merged.dedup.recal.table | BQSR recalibration table. |
01_bam/<id>.rg.merged.dedup.recal.bam | Recalibrated BAM used for variant calling. |
01_bam/*.bai or 01_bam/*.bam.bai | BAM indexes. |
02_varcall/<id>.hc.raw.vcf.gz | Raw VCF from GenotypeGVCFs. |
02_varcall/<id>.hc.raw.vcf.gz.tbi | Tabix index for the raw VCF. |
Conditional VQSR files
These appear only when the run has enough SNPs or indels to build VQSR models.
| File | Meaning |
|---|---|
02_varcall/<id>.hc.snp.recal.vcf.gz | SNP VQSR model output. |
02_varcall/<id>.hc.snp.tranches.txt | SNP VQSR tranche diagnostics. |
02_varcall/<id>.hc.post_snp.vcf.gz | VCF after applying SNP VQSR. |
02_varcall/<id>.hc.indel.recal.vcf.gz | INDEL VQSR model output. |
02_varcall/<id>.hc.indel.tranches.txt | INDEL VQSR tranche diagnostics. |
02_varcall/<id>.hc.vqsr.vcf.gz | VCF after applying SNP and INDEL VQSR. |
If VQSR is skipped because there are too few variants, the final *.hc.QC.vcf.gz is still produced by hard filtering.
WES/WGS Cohort
Applies to pipeline: wes or pipeline: wgs with mode: cohort.
In cohort output filenames, gv is shorthand for GenotypeGVCFs, the GATK
step that converts the GenomicsDB workspace into a joint-genotyped VCF.
Recommended Files
| File | Use |
|---|---|
02_varcall/cohort.gv.QC.vcf.gz | Final filtered cohort VCF. This is the primary joint-genotyped variant file. |
02_varcall/cohort.gv.QC.vcf.gz.tbi | Tabix index for the final cohort VCF. |
Intermediate files
| File | Meaning |
|---|---|
01_genomicsdb/cohort.genomicsdb.<run-id>/ | GenomicsDB workspace used by GenomicsDBImport. |
01_genomicsdb/genomicsdbimport.done | Backend marker showing that GenomicsDB import completed. |
01_genomicsdb/wgs.whole_genome.interval_list | WGS-only interval list derived from the reference dictionary for GenomicsDBImport and GenotypeGVCFs. |
02_varcall/cohort.gv.raw.vcf.gz | Raw cohort VCF from GenotypeGVCFs. |
02_varcall/cohort.gv.raw.vcf.gz.tbi | Tabix index for the raw cohort VCF. |
logs/01_genomicsdbimport.log | GenomicsDB import log. |
logs/02_genotype_gvcfs.log | Cohort genotyping log. |
logs/03_vqsr_and_qc.log | VQSR and final filtering log. |
01_genomicsdb/ is a large intermediate workspace and is excluded from the
audited file inventory in run-report.json. Final VCFs, stats, logs, execution
contracts, and output hashes remain part of the reproducibility surface.
Conditional VQSR files
| File | Meaning |
|---|---|
02_varcall/cohort.snp.recal.vcf.gz | SNP VQSR model output. |
02_varcall/cohort.snp.tranches.txt | SNP VQSR tranche diagnostics. |
02_varcall/cohort.post_snp.vcf.gz | VCF after applying SNP VQSR. |
02_varcall/cohort.indel.recal.vcf.gz | INDEL VQSR model output. |
02_varcall/cohort.indel.tranches.txt | INDEL VQSR tranche diagnostics. |
02_varcall/cohort.vqsr.vcf.gz | VCF after applying SNP and INDEL VQSR. |
mtDNA Single-Sample
Applies to pipeline: mit with mode: single.
Recommended Files
| File | Use |
|---|---|
01_mtoolbox/mit_prioritized_variants.txt | Final prioritized mtDNA variant report with GT, DP, and heteroplasmic fraction columns appended by CBIcall. |
01_mtoolbox/VCF_file.vcf | mtDNA VCF from MToolBox. |
01_mtoolbox/mit.filtered.json | Canonical machine-readable records retained by the browser's HF and population-frequency filters. |
02_browser/<run-id>.html | Standalone interactive mtDNA report with embedded data and browser assets. |
02_browser/README.txt | Local instructions for opening the browser report. |
Intermediate files
| File | Meaning |
|---|---|
01_mtoolbox/<id>-DNA_MIT.bam | Extracted mitochondrial BAM used as MToolBox input. |
01_mtoolbox/<id>-DNA_MIT.bam.bai | BAM index. |
01_mtoolbox/prioritized_variants.txt | Raw MToolBox prioritized variant list before CBIcall appends genotype/depth/HF fields. |
01_mtoolbox/mt_classification_best_results.csv | MToolBox haplogroup/classification output. |
01_mtoolbox/processed_fastq.tar.gz | MToolBox processed FASTQ archive. |
01_mtoolbox/summary_*.txt | MToolBox run summary. |
01_mtoolbox/OUT_*/ | MToolBox working directory with alignment, pileup, coverage, and annotation intermediates. |
mtDNA Cohort
Applies to pipeline: mit with mode: cohort.
The cohort workflow uses the same output directories as mtDNA single-sample mode, but extracts mtDNA BAMs from all matching sibling sample directories before running MToolBox jointly.
Recommended Files
| File | Use |
|---|---|
01_mtoolbox/mit_prioritized_variants.txt | Final joint mtDNA variant report with per-sample GT, DP, and heteroplasmic fraction fields. |
01_mtoolbox/VCF_file.vcf | Joint mtDNA VCF from MToolBox. |
01_mtoolbox/mit.filtered.json | Canonical machine-readable records retained by the browser's HF and population-frequency filters. |
02_browser/<run-id>.html | Standalone interactive cohort report with embedded data and browser assets. |
02_browser/README.txt | Local instructions for opening the browser report. |
Intermediate files
| File | Meaning |
|---|---|
01_mtoolbox/<sample-id>-DNA_MIT.bam | Extracted mitochondrial BAM for each cohort sample. |
01_mtoolbox/<sample-id>-DNA_MIT.bam.bai | BAM index for each extracted mitochondrial BAM. |
01_mtoolbox/prioritized_variants.txt | Raw MToolBox prioritized variant list. |
01_mtoolbox/missing_variants.txt | Temporary variant list used while appending cohort genotype/depth/HF fields. |
01_mtoolbox/OUT_*/ | MToolBox working directories and intermediate outputs. |