Tutorial: Beaconize a Dataset
End-to-end tutorial
Prepare metadata and variants as portable Beacon v2 BFF files.
Start with an XLSX workbook and a VCF, validate each output, and finish with files that can be reviewed or handed to a Beacon implementation.
You need:
- metadata for Beacon entities such as individuals, biosamples, analyses, runs, cohorts, and datasets;
- a VCF/VCF.gz file, or a supported SNP-array TSV/TXT file;
- the correct assembly name for the genomic input.
The toolkit prepares research data and annotations. It is not a medical device and its output must not be used by itself for clinical or medical decisions. See the full disclaimer.
1. Prepare Metadata
Export the workbook shipped with the installed package:
bff-tools validate --template-out metadata.xlsx
The CINECA synthetic workbook is a populated reference for the currently supported schema version.
Workbook headers encode JSON structure:
sex.idcreates a nested object;measures_assayCode.idcreates a one-dimensional array of objects;- values beginning with
[or{are parsed as JSON; - numeric and boolean strings are serialized as JSON numbers and booleans.
Most projects leave the workbook genomicVariations sheet empty because VCF or TSV conversion creates that collection in Step 2. Use --gv only when you intentionally maintain complementary genomic-variation records in the workbook or as a named JSON collection.
Convert and validate the completed workbook:
bff-tools validate -i metadata.xlsx -o bff
🧬 BFF Tools Validator v2.0.13
Build and validate BFF metadata from XLSX, or validate existing BFF JSON
== 🗂 analyses ==
✓ analyses: validation passed (2,504 records)
→ Wrote .../bff/analyses.json
== 🧪 biosamples ==
✓ biosamples: validation passed (2,504 records)
→ Wrote .../bff/biosamples.json
== 👥 cohorts ==
✓ cohorts: validation passed (1 record)
→ Wrote .../bff/cohorts.json
== 📦 datasets ==
✓ datasets: validation passed (1 record)
→ Wrote .../bff/datasets.json
== 🧍 individuals ==
✓ individuals: validation passed (2,504 records)
→ Wrote .../bff/individuals.json
== 🧫 runs ==
✓ runs: validation passed (2,504 records)
→ Wrote .../bff/runs.json
✓ Validation passed; checked 10,018 records
Each valid worksheet becomes a deterministic BFF JSON collection in the output directory. Use --no-color or --no-emoji when plain output is required for a log processor.
Diagnose a Failed Workbook Row
The report names the collection, the actual workbook row, and the path in the generated JSON. It is normal to validate repeatedly while mapping a local data model.
The ontology identifier is not a CURIE
individuals: 1 validation issue(s)
row 2: ethnicity.id: 'European' does not match '^\w[^:]+:.+$'
The .id cell needs a real ontology identifier in PREFIX:TERM form. Put human-readable text in the matching .label cell. Do not invent an identifier merely to satisfy the pattern.
A required identifier is missing
individuals: 1 validation issue(s)
row 2: $: 'id' is a required property
Add a stable, unique entity identifier to the id cell on workbook row 2. The row number is the Excel row, including the header row.
A oneOf value matches multiple schema alternatives
row 2: diseases.0.ageOfOnset: ... is valid under each of ...
oneOf requires exactly one representation. Inspect the generated object and remove mixed age, range, date, ontology, quantity, or measurement alternatives. Some Beacon v2 branches overlap; --ignore-validation may expose the intermediate JSON for diagnosis, but it does not turn that output into a validation pass.
Values beginning with [ or { must be valid JSON. Unicode is supported, but pasted non-breaking spaces, typographic quotes, and invisible control characters can make a cell difficult to diagnose. Correct the workbook and rerun without --ignore-validation before using the output.
A successful metadata run normally writes six collections: analyses.json, biosamples.json, cohorts.json, datasets.json, individuals.json, and runs.json. Empty worksheets are omitted.
2. Convert Variants
Most raw VCFs should use the fully configured annotation workflow:
export BFF_TOOLS_DATA=/absolute/path/to/beacon2-cbi-tools-data
bff-tools vcf \
-i cohort.vcf.gz \
--genome hg38 \
--dataset-id cohort-1 \
--annotate \
-o cohort-bff
This normalizes the VCF and applies SnpEff, dbNSFP, ClinVar, and COSMIC resources before the production converter maps common fields, genotypes, ANN consequences, prediction data, and clinical interpretations.
--dataset-id must identify the same dataset represented in datasets.json and referenced by the connected metadata. For multi-sample VCFs, sample names become biosampleId values in caseLevelData; verify that they match your biosample identifiers.
The production mapping targets DNA-sequencing SNVs and nucleotide insertions/deletions from single-sample or multi-sample VCFs. Symbolic structural and copy-number alleles are currently skipped; see Supported Data before processing an SV-focused callset.
The full stdout lists the arguments, resolved configuration, and input parameters. A successful compact run ends with:
Starting BFF-Tools
Pipeline execution begins now
🧬 VCF2BFF
🌐 BFF2HTML
BFF-Tools Finished
Status => OK
Runtime => 1s
Conversion writes compressed BFF genomic variations plus log.json, generated stage scripts, and stage logs. Runtime depends on input size, annotations, sample count, CPU, and storage.
The assembly must match the input. b37 is normalized to hs37; hg19, hs37, and hg38 remain distinct because contig conventions and coordinates can differ.
Already Annotated Input
Skip the external stages only when the VCF already contains a compatible SnpEff ANN header and annotations. dbNSFP and ClinVar fields are still strongly recommended for complete BFF records:
bff-tools vcf \
-i cohort.annotated.vcf.gz \
--genome hg38 \
--dataset-id cohort-1 \
--no-annotate \
-o cohort-bff
On a 6-core ARM64 Linux workstation, an end-to-end run completed in 13 minutes 52 seconds. It normalized 1,103,547 source records into 1,110,240 records, applied SnpEff, dbNSFP, ClinVar, and COSMIC annotations, produced 1,109,368 BFF records for 2,504 samples, and generated a standalone report containing 14,305 panel-matched variants.
Measured separately on the same workstation, the single-process converter processed the already annotated VCF in 1 minute 47 seconds with about 20 MB peak RAM. That VCF uses FORMAT=GT; multi-field sample values such as GT:DP:AD require additional parsing and may take longer. Browser generation took 44 seconds with about 22 MB peak RAM. These are observed timings rather than hardware-independent guarantees; CPU, storage, compression, reference versions, and annotation settings materially affect runtime.
Resource versions materially affect output and must be recorded with every annotation-enabled run. Complete setup and the packaged compact integration test are documented under Annotation Data.
Diagnose a Failed VCF Run
Start with the log path printed by bff-tools; the generated stage script records the exact command that failed.
The VCF has no usable SnpEff ANN header
Raw VCF input must run annotation, which is enabled by default. This error usually means --no-annotate or annotate: false was used on an unannotated file. Remove that override and select the annotation data through BFF_TOOLS_DATA.
bcftools reports missing contigs or reference mismatches
Compare input contigs with the selected FASTA index:
bcftools query -f '%CHROM\n' cohort.vcf.gz | head
cut -f1 /path/to/reference.fa.gz.fai | head
Select resources with matching coordinates and names. chr22 versus 22 is a contig convention; changing the text does not perform a liftover or make hg19 and hs37 interchangeable.
SnpEff reports a missing database
The local genome database is missing or BFF_TOOLS_DATA does not name the bundle path visible inside the container. The standard database directory is $BFF_TOOLS_DATA/databases/snpeff/v5.0; the generated command passes it with -dataDir and disables downloads.
bcftools names a malformed INFO tag
Fix the producer or VCF header when possible. If the named tag is disposable, remove only that tag with bcftools annotate -x INFO/TAG, preserve the original, and record the transformation. The historical INFO/IDREP command is retained in the FAQ.
The requested output directory already exists
Runs never overwrite an existing project directory. Choose a new path with -o, or archive and move the previous run before retrying.
Annotation retains normalized, SnpEff, dbNSFP, ClinVar, and COSMIC VCF intermediates plus the final BFF JSON. Reserve up to 10 times the compressed input VCF size as a planning estimate, in addition to the annotation bundle. Preserve the final annotated VCF, logs, scripts, checksums, and resource versions before deleting intermediates.
SNP-Array Input
For a supported TSV/TXT export:
bff-tools tsv \
-i genotypes.txt.gz \
--sample-id sample-1 \
--genome hg19 \
--dataset-id cohort-1
TSV conversion requires bcftools, the matching reference FASTA, and the full annotation profile. It cannot use --no-annotate because the generated VCF has no SnpEff ANN data.
3. Validate Genomic Output
The primary variant output is:
cohort-bff/vcf/genomicVariationsVcf.json.gz
Validate its records without loading the whole collection into memory:
bff-tools validate \
-i cohort-bff/vcf/genomicVariationsVcf.json.gz \
--gv-vcf
4. Inspect and Hand Off
Generate the standalone BFF Tools Browser during conversion:
bff-tools vcf -i cohort.vcf.gz \
--genome hg38 --dataset-id cohort-1 \
--browser -o cohort-bff-browser
Open the generated HTML file directly. The adjacent browser/README.txt records the exact filename and an optional local HTTP-server command. The report includes searching, sorting, column controls, pagination for large tables, gene panels, a structured variant inspector, and assembly-aware links to external variant databases.
Click any row to open the variant inspector without leaving the table. It groups the clinical interpretation, variant definition, external evidence links, matched panels, and carrier genotypes while preserving the current filters and page. Categorical values retain the same color semantics used in the table.
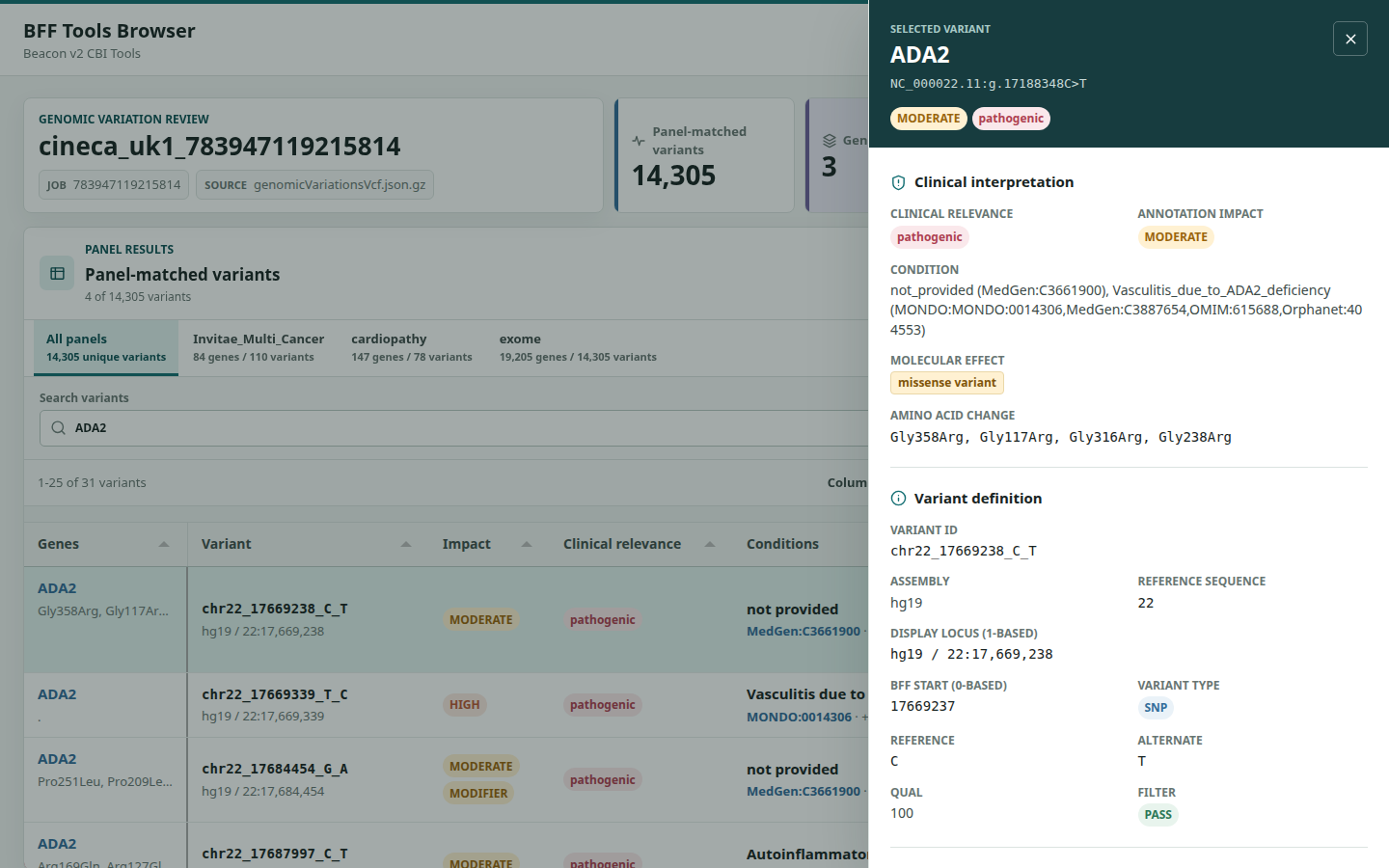
Before handing the BFF files to a Beacon implementation, confirm:
- metadata and genomic collections pass schema validation;
- the selected assembly and contig convention match the source data;
- expected sample, variant, and entity counts are present;
- a representative set of variants and annotations has been inspected;
log.json, parameter YAML, annotation resource versions, and source checksums are retained.
5. Connect to a Beacon Backend
bff-tools deliberately stops at portable BFF files. A backend can consume them without being bundled into the data-preparation application.
- Follow MongoDB Import for indexes, repeatable upserts, and large streamed collections.
- See EGA Beacon v2 PI API or Progenetix bycon for complete Beacon server implementations.
MongoDB clients are external to the application, Docker image, and current r3 annotation bundle. Install them separately only when required by the downstream Beacon implementation.
Continue with Validation and Reproducibility for the release checklist or the FAQ for failed runs.