Using Pheno-Ranker with OMIM Data
This guide explains how to use Pheno-Ranker with data from the OMIM Database.
Data Download
Starting from version 1.02, Pheno-Ranker includes disease-based cohorts: one for the OMIM Database and another for the ORPHAnet Database.
We provide two data exchange formats, PXF (Phenotype Exchange Format) and BFF (Beacon Friendly Format).
For more details on how these cohorts were created, refer to this repository. If you use this data in your research, please check the citation section.
If you installed the code via GitHub or Docker, the disease-based cohorts should already be available at share/diseases/hpo/. Otherwise, follow these steps to download them. Ensure that wget and jq are installed:
sudo apt install wget jq
We assume that you have R installed and can install its required packages.
wget https://raw.githubusercontent.com/CNAG-Biomedical-Informatics/pheno-ranker/refs/heads/main/share/diseases/hpo/omim.pxf.json.gz
Here, we will use the PXF version of the disease-based cohort.
Analytics
zcat omim.pxf.json.gz | jq 'length'
The cohort consists of 6,471 diseases. Each disease is represented as a PXF containing a list of phenotypicTerms.
From this point onward, we assume that the pheno-ranker directory is located at ../. Please adjust the path accordingly.
To analyze the distribution of terms across patients, we will use a Perl script.
See Perl code
#!/usr/bin/perl
use strict;
use warnings;
use JSON::XS;
# Read from STDIN only
if (-t STDIN) { # Check if STDIN is empty (no piped input)
print STDERR "Usage: zcat input.json.gz | $0\n";
print STDERR " cat input.json | $0\n";
exit 1;
}
# Read the entire JSON content from STDIN
my $json_text = do {
local $/;
<STDIN>;
};
# Decode JSON
my $json = JSON::XS->new->utf8->decode($json_text);
# Ensure the decoded JSON is an array reference
die "Input JSON is not an array.\n" unless ref($json) eq 'ARRAY';
# Print CSV header
print "key,count\n";
# Iterate through each object in the array
foreach my $obj (@$json) {
# Ensure the current element is an object with an 'id' field
next unless ref($obj) eq 'HASH' && exists $obj->{id};
my $id = $obj->{id};
my $count = 0;
# Check if 'phenotypicFeatures' exists and is an array
if (exists $obj->{phenotypicFeatures} && ref($obj->{phenotypicFeatures}) eq 'ARRAY') {
$count = scalar @{ $obj->{phenotypicFeatures} };
}
# Print the CSV line
print "\"$id\",$count\n";
}
zcat omim.pxf.json.gz | ./count_phenotypicFeatures.pl > counts.csv
Now, we will use R to plot a histogram.
See R code
# Load required packages
library(ggplot2)
# Read the CSV file into a data frame
df <- read.csv("counts.csv")
# Calculate the average and median counts
average_count <- mean(df$count)
median_count <- median(df$count)
# Generate the histogram and add vertical lines for the average and median
plot <- ggplot(df, aes(x = count)) +
geom_histogram(binwidth = 1, fill = "steelblue", color = "black") +
geom_vline(aes(xintercept = average_count), color = "black", linetype = "dashed", linewidth = 1) +
geom_vline(aes(xintercept = median_count), color = "red", linetype = "dashed", linewidth = 1) +
labs(title = "Distribution of Element Counts per Object",
x = "Number of Elements",
y = "Frequency") +
theme_minimal() +
annotate("text", x = average_count + 0.5, y = Inf, label = paste("Mean:", round(average_count, 2)), vjust = 2) +
annotate("text", x = median_count - 0.5, y = Inf, label = paste("Median:", round(median_count, 2)), vjust = 2, color = "red")
# Save the plot as a PNG file
ggsave("histogram_with_mean_median.png", plot = plot, width = 8, height = 6, dpi = 300)
# Print confirmation
cat("Histogram saved as histogram_with_mean_median.png\n")
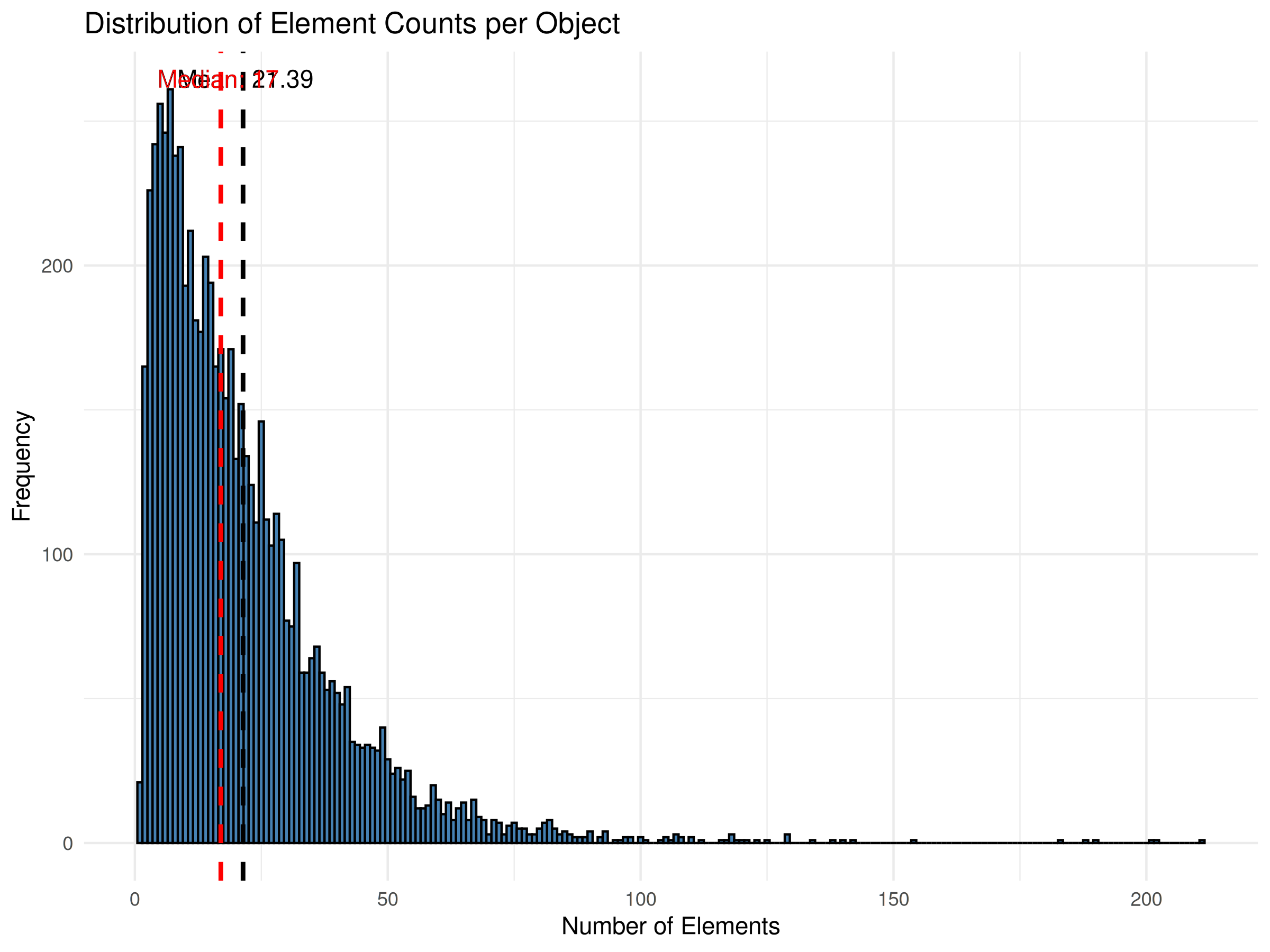
Based on the median, the Completeness of the Phenopackets Corpus is approximately 71% (12/17).
Cohort Mode
We begin with a simple calculation in cohort mode. In the run, we will also export intermediate files using --e omim for later use. From now on, we will focus on phenotypicFeatures terms, which we aim to use for patient classification.
Ensure that the pheno-ranker directory is set correctly.
time ../pheno-ranker/bin/pheno-ranker -r omim.pxf.json.gz --include-terms phenotypicFeatures --max-matrix-records-in-ram 10000 -e omim
This calculation takes approximately 1 min 40 sec (1 core @ Apple M2 Pro). The --max-matrix-records-in-ram 10000 flag increases efficiency by using RAM, making the process 2x faster.
The default output is a dense matrix.txt, which is useful for the included R scripts. If your downstream workflow accepts sparse matrices, use Matrix Market output instead:
time ../pheno-ranker/bin/pheno-ranker -r omim.pxf.json.gz --include-terms phenotypicFeatures --matrix-format mtx -o omim.mtx -e omim
Graph exports can also become large. Use --graph-max-weight with Hamming distance to keep close pairs, or --graph-min-weight with Jaccard to keep highly similar pairs.
Since a 6,471 × 6,471 matrix is too large for a heatmap, we will use multidimensional scaling (mds.R script).
Rscript ../pheno-ranker/share/r/mds.R
This computation takes about 5 minutes (1 core @ Apple M2 Pro).
See R code
library(ggplot2)
library(ggrepel)
# Read in the input file as a matrix
data <- as.matrix(read.table("matrix.txt", header = TRUE, row.names = 1, check.names = FALSE))
# Calculate distance matrix
#d <- dist(data)
#d <- 1 - data # J-similarity to J-distance
# Perform multidimensional scaling
#fit <- cmdscale(d, eig=TRUE, k=2)
fit <- cmdscale(data, eig=TRUE, k=2)
# Extract (x, y) coordinates of multidimensional scaling
x <- fit$points[,1]
y <- fit$points[,2]
# Create data frame
df <- data.frame(x, y, label=row.names(data))
# Save image
png(filename = "mds.png", width = 1000, height = 1000,
units = "px", pointsize = 12, bg = "white", res = NA)
# Create scatter plot
ggplot(df, aes(x, y, label = label)) +
geom_point() +
geom_text_repel(size = 5, # Adjust the size of the text
box.padding = 0.2, # Adjust the padding around the text
max.overlaps = 10) + # Change the maximum number of overlaps
labs(title = "Multidimensional Scaling Results",
x = "Hamming Distance MDS Coordinate 1",
y = "Hamming Distance MDS Coordinate 2") + # Add title and axis labels
theme(
plot.title = element_text(size = 30, face = "bold", hjust = 0.5),
axis.title = element_text(size = 25),
axis.text = element_text(size = 15))
#dev.off()
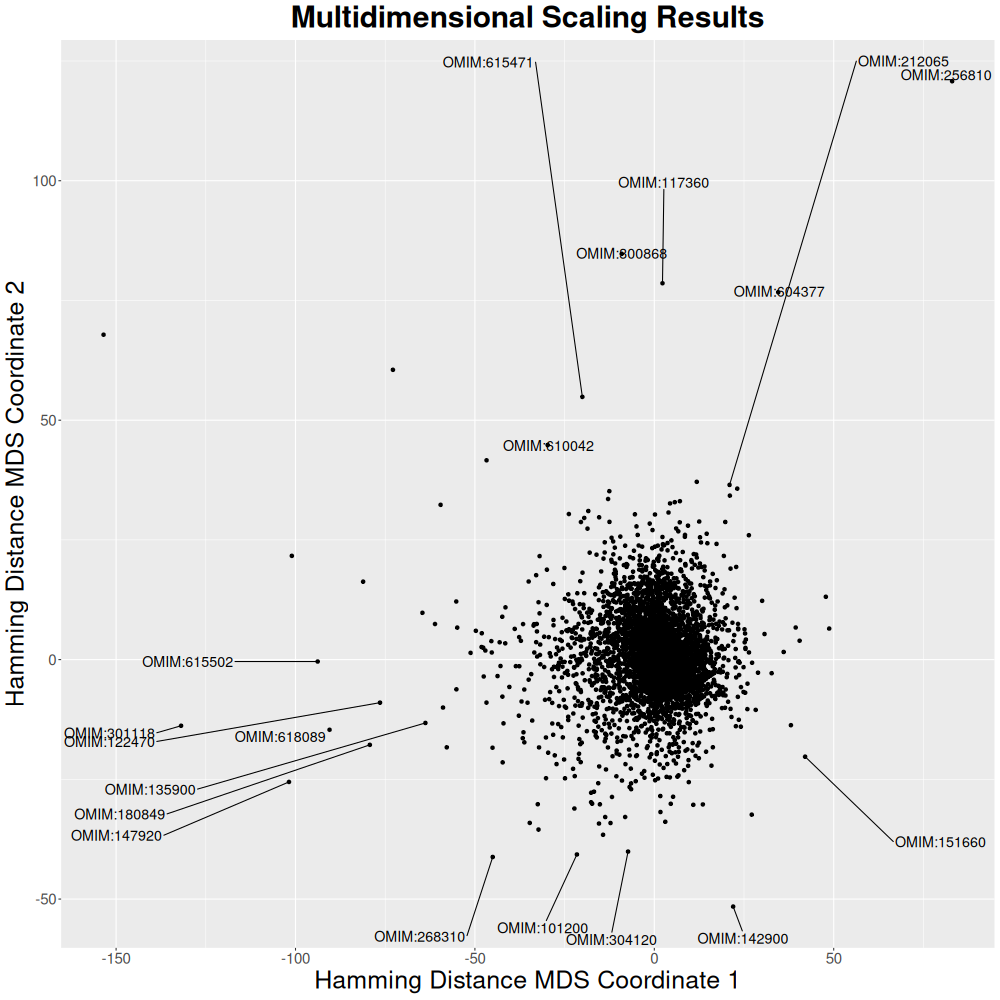
Graph Representation
To generate a graph visualization, we use a subset of 50 diseases for faster execution.
zcat omim.pxf.json.gz | jq -c '.[]' | shuf -n 50 | jq -s '.' > omim_small.json
../pheno-ranker/bin/pheno-ranker -r omim_small.json -include-terms phenotypicFeatures --cytoscape-json omim_cytoscape.json --graph-max-weight 10
Loading graph...
Patient Mode
We will attempt to match patient PMID_35344616_A2 from the Phenopackets Corpus to its disease (OMIM:268310) in OMIM. See the Phenopackets Corpus tutorial for instructions on obtaining a PXF file for the patient.
Sorting by Jaccard index is recommended, as data completeness is below 30%. From the top three, selecting by INTERSECT-RATE(%) ranks the match at 1.
../pheno-ranker/bin/pheno-ranker -r omim.pxf.json.gz -t PMID_35344616_A2.json -include-terms phenotypicFeatures -sort-by jaccard -max-out 5
| RANK | REFERENCE(ID) | TARGET(ID) | FORMAT | LENGTH | WEIGHTED | HAMMING-DISTANCE | DISTANCE-Z-SCORE | DISTANCE-P-VALUE | DISTANCE-Z-SCORE(RAND) | JACCARD-INDEX | JACCARD-Z-SCORE | JACCARD-P-VALUE | REFERENCE-VARS | TARGET-VARS | INTERSECT | INTERSECT-RATE(%) | COMPLETENESS(%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | OMIM:180700 | PMID_35344616_A2 | PXF | 77 | False | 52 | 0.111 | 0.5440913 | 3.0769 | 0.325 | 10.784 | 0.0000000 | 71 | 31 | 25 | 80.65 | 35.21 |
| 2 | OMIM:616894 | PMID_35344616_A2 | PXF | 64 | False | 44 | -0.397 | 0.3457285 | 3.0000 | 0.312 | 10.359 | 0.0000000 | 53 | 31 | 20 | 64.52 | 37.74 |
| 3 | OMIM:268310 | PMID_35344616_A2 | PXF | 107 | False | 76 | 1.634 | 0.9488308 | 4.3503 | 0.290 | 9.563 | 0.0000000 | 107 | 31 | 31 | 100.00 | 28.97 |
| 4 | OMIM:618529 | PMID_35344616_A2 | PXF | 53 | False | 39 | -0.714 | 0.2375690 | 3.4340 | 0.264 | 8.670 | 0.0000000 | 36 | 31 | 14 | 45.16 | 38.89 |
| 5 | OMIM:616331 | PMID_35344616_A2 | PXF | 84 | False | 64 | 0.872 | 0.8084460 | 4.8008 | 0.238 | 7.759 | 0.0000000 | 73 | 31 | 20 | 64.52 | 27.40 |
This process takes ~10 seconds. For a much faster approach using precomputed data, run:
../pheno-ranker/bin/pheno-ranker -prp omim -t PMID_35344616_A2.json -include-terms phenotypicFeatures -sort-by jaccard -max-out 5
With precomputed data, the calculation takes only 0.5 seconds while yielding identical results.
All-to-All Precomputed Similarity
We have precomputed pairwise similarity scores for every OMIM entry and stored them in a backend database. The proof-of-concept web app lets you enter any OMIM ID (TAR-UUID) or filter by any database column and retrieve the top five most similar diseases.
Go to the Pheno-Ranker Use Cases Playground.
The current interface is experimental and intended to demonstrate the workflow.
At this time, we don’t display the matched HPO terms—this feature will be available in a future release. However, each result includes a clickable link to the corresponding OMIM entry, so you can quickly review the detailed information.
Citation
If you use this information in your research, please cite the following: