Cohort Mode
Cohort mode performs an all-vs-all comparison of records in one or more cohorts. Each record is flattened, encoded as a binary vector, and compared with either Hamming distance or the Jaccard index.
Use cohort mode when you want to explore the structure of a cohort, compare multiple cohorts, identify clusters, run dimensionality reduction, or export a graph for network analysis.
pheno-ranker -r cohort.jsonmatrix.txtWhat You Get
matrix.txt: the default dense pairwise comparison matrix.graph.json: an optional Cytoscape-compatible graph when--cytoscape-jsonis used.graph_stats.txt: optional graph summary statistics when--graph-statsis used.export.*.json: optional intermediate hashes, vectors, and coverage statistics when--exportis used.matrix.mtx: optional sparse Matrix Market output for large matrix workflows.
For target-to-reference ranking instead, see Patient Mode. For custom categorical records, see the Generic JSON tutorial.
Usage
The examples below show common cohort-mode command-line patterns. For the complete CLI reference, see Usage.
- Intra-cohort
- Inter-cohort
For this example, we use individuals.json, a JSON array with 36 patients. The goal is to compare every patient against every other patient in the file.
First, we will download the file:
wget https://raw.githubusercontent.com/CNAG-Biomedical-Informatics/pheno-ranker/refs/heads/main/t/data/individuals.json
Now run Pheno-Ranker:
pheno-ranker -r individuals.json
More input examples
You can find more input examples here.
This process generates a matrix.txt file, containing the results of 36 x 36 pairwise comparisons, calculated using the Hamming distance metric.
See matrix.txt
| 107:week_0_arm_1 | 107:week_2_arm_1 | 107:week_14_arm_1 | 125:week_0_arm_1 | 125:week_2_arm_1 | 125:week_14_arm_1 | 125:week_26_arm_1 | 125:week_52_arm_1 | 125:week_78_arm_1 | 215:week_0_arm_1 | 215:week_2_arm_1 | 215:week_14_arm_1 | 215:week_26_arm_1 | 215:week_52_arm_1 | 215:week_78_arm_1 | 257:week_0_arm_1 | 257:week_2_arm_1 | 257:week_14_arm_1 | 257:week_26_arm_1 | 275:week_0_arm_1 | 275:week_2_arm_1 | 275:week_14_arm_1 | 275:week_52_arm_1 | 305:week_0_arm_1 | 305:week_26_arm_1 | 305:week_52_arm_1 | 365:week_0_arm_1 | 365:week_2_arm_1 | 365:week_14_arm_1 | 365:week_26_arm_1 | 365:week_52_arm_1 | 527:week_0_arm_1 | 527:week_2_arm_1 | 527:week_14_arm_1 | 527:week_26_arm_1 | 527:week_52_arm_1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 107:week_0_arm_1 | 0 | 24 | 23 | 6 | 23 | 23 | 24 | 43 | 40 | 16 | 27 | 29 | 27 | 49 | 32 | 29 | 45 | 45 | 50 | 14 | 25 | 30 | 51 | 18 | 26 | 45 | 20 | 25 | 26 | 30 | 45 | 24 | 24 | 23 | 32 | 43 |
| 107:week_2_arm_1 | 24 | 0 | 3 | 22 | 3 | 3 | 2 | 23 | 18 | 30 | 7 | 9 | 7 | 29 | 10 | 47 | 25 | 25 | 28 | 30 | 5 | 10 | 31 | 32 | 4 | 25 | 34 | 5 | 6 | 8 | 25 | 42 | 2 | 3 | 10 | 23 |
| 107:week_14_arm_1 | 23 | 3 | 0 | 21 | 2 | 2 | 3 | 22 | 19 | 29 | 6 | 8 | 6 | 28 | 11 | 46 | 24 | 24 | 29 | 29 | 4 | 9 | 30 | 31 | 5 | 24 | 33 | 4 | 5 | 9 | 24 | 41 | 3 | 2 | 11 | 22 |
| 125:week_0_arm_1 | 6 | 22 | 21 | 0 | 21 | 21 | 22 | 41 | 38 | 14 | 25 | 27 | 25 | 47 | 30 | 29 | 43 | 43 | 48 | 12 | 23 | 28 | 49 | 16 | 24 | 43 | 18 | 23 | 24 | 28 | 43 | 24 | 22 | 21 | 30 | 41 |
| 125:week_2_arm_1 | 23 | 3 | 2 | 21 | 0 | 2 | 3 | 22 | 19 | 29 | 6 | 8 | 6 | 28 | 11 | 46 | 24 | 24 | 29 | 29 | 4 | 9 | 30 | 31 | 5 | 24 | 33 | 4 | 5 | 9 | 24 | 41 | 3 | 2 | 11 | 22 |
| 125:week_14_arm_1 | 23 | 3 | 2 | 21 | 2 | 0 | 3 | 22 | 19 | 29 | 6 | 8 | 6 | 28 | 11 | 46 | 24 | 24 | 29 | 29 | 4 | 9 | 30 | 31 | 5 | 24 | 33 | 4 | 5 | 9 | 24 | 41 | 3 | 2 | 11 | 22 |
| 125:week_26_arm_1 | 24 | 2 | 3 | 22 | 3 | 3 | 0 | 23 | 18 | 30 | 7 | 9 | 7 | 29 | 10 | 47 | 25 | 25 | 28 | 30 | 5 | 10 | 31 | 32 | 4 | 25 | 34 | 5 | 6 | 8 | 25 | 42 | 2 | 3 | 10 | 23 |
| 125:week_52_arm_1 | 43 | 23 | 22 | 41 | 22 | 22 | 23 | 0 | 7 | 49 | 26 | 28 | 26 | 8 | 15 | 26 | 4 | 4 | 9 | 49 | 24 | 29 | 10 | 51 | 25 | 4 | 53 | 24 | 25 | 29 | 4 | 21 | 23 | 22 | 15 | 2 |
| 125:week_78_arm_1 | 40 | 18 | 19 | 38 | 19 | 19 | 18 | 7 | 0 | 46 | 23 | 25 | 23 | 13 | 10 | 31 | 9 | 9 | 12 | 46 | 21 | 26 | 15 | 48 | 20 | 9 | 50 | 21 | 22 | 24 | 9 | 26 | 18 | 19 | 10 | 7 |
| 215:week_0_arm_1 | 16 | 30 | 29 | 14 | 29 | 29 | 30 | 49 | 46 | 0 | 33 | 27 | 33 | 43 | 38 | 37 | 51 | 51 | 56 | 12 | 31 | 34 | 45 | 22 | 32 | 51 | 18 | 31 | 30 | 36 | 51 | 34 | 30 | 29 | 38 | 49 |
| 215:week_2_arm_1 | 27 | 7 | 6 | 25 | 6 | 6 | 7 | 26 | 23 | 33 | 0 | 12 | 2 | 32 | 15 | 50 | 28 | 28 | 33 | 33 | 8 | 13 | 34 | 35 | 9 | 28 | 29 | 8 | 9 | 13 | 28 | 45 | 7 | 6 | 15 | 26 |
| 215:week_14_arm_1 | 29 | 9 | 8 | 27 | 8 | 8 | 9 | 28 | 25 | 27 | 12 | 0 | 12 | 26 | 17 | 50 | 30 | 30 | 35 | 23 | 10 | 13 | 28 | 37 | 11 | 30 | 31 | 10 | 9 | 15 | 30 | 47 | 9 | 8 | 17 | 28 |
| 215:week_26_arm_1 | 27 | 7 | 6 | 25 | 6 | 6 | 7 | 26 | 23 | 33 | 2 | 12 | 0 | 32 | 15 | 50 | 28 | 28 | 33 | 33 | 8 | 13 | 34 | 35 | 9 | 28 | 29 | 8 | 9 | 13 | 28 | 45 | 7 | 6 | 15 | 26 |
| 215:week_52_arm_1 | 49 | 29 | 28 | 47 | 28 | 28 | 29 | 8 | 13 | 43 | 32 | 26 | 32 | 0 | 21 | 30 | 10 | 10 | 15 | 47 | 30 | 33 | 4 | 57 | 31 | 10 | 55 | 30 | 29 | 35 | 10 | 27 | 29 | 28 | 21 | 8 |
| 215:week_78_arm_1 | 32 | 10 | 11 | 30 | 11 | 11 | 10 | 15 | 10 | 38 | 15 | 17 | 15 | 21 | 0 | 39 | 17 | 17 | 20 | 38 | 13 | 18 | 23 | 40 | 12 | 17 | 42 | 13 | 14 | 16 | 17 | 34 | 10 | 11 | 2 | 15 |
| 257:week_0_arm_1 | 29 | 47 | 46 | 29 | 46 | 46 | 47 | 26 | 31 | 37 | 50 | 50 | 50 | 30 | 39 | 0 | 24 | 24 | 29 | 31 | 44 | 47 | 28 | 37 | 45 | 24 | 37 | 44 | 43 | 49 | 24 | 7 | 47 | 46 | 39 | 26 |
| 257:week_2_arm_1 | 45 | 25 | 24 | 43 | 24 | 24 | 25 | 4 | 9 | 51 | 28 | 30 | 28 | 10 | 17 | 24 | 0 | 2 | 7 | 47 | 22 | 27 | 8 | 49 | 23 | 2 | 51 | 22 | 23 | 27 | 2 | 23 | 25 | 24 | 17 | 4 |
| 257:week_14_arm_1 | 45 | 25 | 24 | 43 | 24 | 24 | 25 | 4 | 9 | 51 | 28 | 30 | 28 | 10 | 17 | 24 | 2 | 0 | 7 | 47 | 22 | 27 | 8 | 49 | 23 | 2 | 51 | 22 | 23 | 27 | 2 | 23 | 25 | 24 | 17 | 4 |
| 257:week_26_arm_1 | 50 | 28 | 29 | 48 | 29 | 29 | 28 | 9 | 12 | 56 | 33 | 35 | 33 | 15 | 20 | 29 | 7 | 7 | 0 | 52 | 27 | 32 | 13 | 46 | 26 | 7 | 56 | 27 | 28 | 22 | 7 | 28 | 28 | 29 | 20 | 9 |
| 275:week_0_arm_1 | 14 | 30 | 29 | 12 | 29 | 29 | 30 | 49 | 46 | 12 | 33 | 23 | 33 | 47 | 38 | 31 | 47 | 47 | 52 | 0 | 27 | 30 | 45 | 18 | 28 | 47 | 12 | 27 | 26 | 32 | 47 | 32 | 30 | 29 | 38 | 49 |
| 275:week_2_arm_1 | 25 | 5 | 4 | 23 | 4 | 4 | 5 | 24 | 21 | 31 | 8 | 10 | 8 | 30 | 13 | 44 | 22 | 22 | 27 | 27 | 0 | 7 | 28 | 29 | 3 | 22 | 31 | 2 | 3 | 7 | 22 | 43 | 5 | 4 | 13 | 24 |
| 275:week_14_arm_1 | 30 | 10 | 9 | 28 | 9 | 9 | 10 | 29 | 26 | 34 | 13 | 13 | 13 | 33 | 18 | 47 | 27 | 27 | 32 | 30 | 7 | 0 | 31 | 34 | 8 | 27 | 34 | 7 | 6 | 12 | 27 | 48 | 10 | 9 | 18 | 29 |
| 275:week_52_arm_1 | 51 | 31 | 30 | 49 | 30 | 30 | 31 | 10 | 15 | 45 | 34 | 28 | 34 | 4 | 23 | 28 | 8 | 8 | 13 | 45 | 28 | 31 | 0 | 55 | 29 | 8 | 53 | 28 | 27 | 33 | 8 | 29 | 31 | 30 | 23 | 10 |
| 305:week_0_arm_1 | 18 | 32 | 31 | 16 | 31 | 31 | 32 | 51 | 48 | 22 | 35 | 37 | 35 | 57 | 40 | 37 | 49 | 49 | 46 | 18 | 29 | 34 | 55 | 0 | 30 | 49 | 22 | 29 | 30 | 26 | 49 | 36 | 32 | 31 | 40 | 51 |
| 305:week_26_arm_1 | 26 | 4 | 5 | 24 | 5 | 5 | 4 | 25 | 20 | 32 | 9 | 11 | 9 | 31 | 12 | 45 | 23 | 23 | 26 | 28 | 3 | 8 | 29 | 30 | 0 | 23 | 32 | 3 | 4 | 6 | 23 | 44 | 4 | 5 | 12 | 25 |
| 305:week_52_arm_1 | 45 | 25 | 24 | 43 | 24 | 24 | 25 | 4 | 9 | 51 | 28 | 30 | 28 | 10 | 17 | 24 | 2 | 2 | 7 | 47 | 22 | 27 | 8 | 49 | 23 | 0 | 51 | 22 | 23 | 27 | 2 | 23 | 25 | 24 | 17 | 4 |
| 365:week_0_arm_1 | 20 | 34 | 33 | 18 | 33 | 33 | 34 | 53 | 50 | 18 | 29 | 31 | 29 | 55 | 42 | 37 | 51 | 51 | 56 | 12 | 31 | 34 | 53 | 22 | 32 | 51 | 0 | 31 | 30 | 36 | 51 | 38 | 34 | 33 | 42 | 53 |
| 365:week_2_arm_1 | 25 | 5 | 4 | 23 | 4 | 4 | 5 | 24 | 21 | 31 | 8 | 10 | 8 | 30 | 13 | 44 | 22 | 22 | 27 | 27 | 2 | 7 | 28 | 29 | 3 | 22 | 31 | 0 | 3 | 7 | 22 | 43 | 5 | 4 | 13 | 24 |
| 365:week_14_arm_1 | 26 | 6 | 5 | 24 | 5 | 5 | 6 | 25 | 22 | 30 | 9 | 9 | 9 | 29 | 14 | 43 | 23 | 23 | 28 | 26 | 3 | 6 | 27 | 30 | 4 | 23 | 30 | 3 | 0 | 8 | 23 | 44 | 6 | 5 | 14 | 25 |
| 365:week_26_arm_1 | 30 | 8 | 9 | 28 | 9 | 9 | 8 | 29 | 24 | 36 | 13 | 15 | 13 | 35 | 16 | 49 | 27 | 27 | 22 | 32 | 7 | 12 | 33 | 26 | 6 | 27 | 36 | 7 | 8 | 0 | 27 | 48 | 8 | 9 | 16 | 29 |
| 365:week_52_arm_1 | 45 | 25 | 24 | 43 | 24 | 24 | 25 | 4 | 9 | 51 | 28 | 30 | 28 | 10 | 17 | 24 | 2 | 2 | 7 | 47 | 22 | 27 | 8 | 49 | 23 | 2 | 51 | 22 | 23 | 27 | 0 | 23 | 25 | 24 | 17 | 4 |
| 527:week_0_arm_1 | 24 | 42 | 41 | 24 | 41 | 41 | 42 | 21 | 26 | 34 | 45 | 47 | 45 | 27 | 34 | 7 | 23 | 23 | 28 | 32 | 43 | 48 | 29 | 36 | 44 | 23 | 38 | 43 | 44 | 48 | 23 | 0 | 42 | 41 | 34 | 21 |
| 527:week_2_arm_1 | 24 | 2 | 3 | 22 | 3 | 3 | 2 | 23 | 18 | 30 | 7 | 9 | 7 | 29 | 10 | 47 | 25 | 25 | 28 | 30 | 5 | 10 | 31 | 32 | 4 | 25 | 34 | 5 | 6 | 8 | 25 | 42 | 0 | 3 | 10 | 23 |
| 527:week_14_arm_1 | 23 | 3 | 2 | 21 | 2 | 2 | 3 | 22 | 19 | 29 | 6 | 8 | 6 | 28 | 11 | 46 | 24 | 24 | 29 | 29 | 4 | 9 | 30 | 31 | 5 | 24 | 33 | 4 | 5 | 9 | 24 | 41 | 3 | 0 | 11 | 22 |
| 527:week_26_arm_1 | 32 | 10 | 11 | 30 | 11 | 11 | 10 | 15 | 10 | 38 | 15 | 17 | 15 | 21 | 2 | 39 | 17 | 17 | 20 | 38 | 13 | 18 | 23 | 40 | 12 | 17 | 42 | 13 | 14 | 16 | 17 | 34 | 10 | 11 | 0 | 15 |
| 527:week_52_arm_1 | 43 | 23 | 22 | 41 | 22 | 22 | 23 | 2 | 7 | 49 | 26 | 28 | 26 | 8 | 15 | 26 | 4 | 4 | 9 | 49 | 24 | 29 | 10 | 51 | 25 | 4 | 53 | 24 | 25 | 29 | 4 | 21 | 23 | 22 | 15 | 0 |
Defining the similarity metric
Use --similarity-metric-cohort to choose the cohort metric. The default value is hamming; the alternative is jaccard.
pheno-ranker -r individuals.json --similarity-metric-cohort jaccard
Sparse Matrix Market output
By default, cohort mode writes a dense tab-separated matrix (matrix.txt). For large cohorts, you can instead write a sparse Matrix Market coordinate file:
pheno-ranker -r individuals.json --matrix-format mtx -o matrix.mtx
The mtx format stores one triangle of the symmetric matrix and writes only non-zero values. It is always RAM-light and does not use the dense in-memory matrix cache controlled by --max-matrix-records-in-ram.
The Matrix Market file includes comment lines mapping 1-based matrix indexes back to individual IDs:
% id 1 107:week_0_arm_1
% id 2 107:week_2_arm_1
Matrix output and Cytoscape graph output are generated independently. This means --matrix-format mtx can be combined with --cytoscape-json.
Exporting intermediate files
It is possible to export all intermediate files, as well as a file indicating coverage, with --export (--e).
Examples:
pheno-ranker -r individuals.json --export
pheno-ranker -r individuals.json --export my_fav_id # choose a prefix
The intermediate files can be used for further processing (e.g., import to a database; see FAQs) or to make informed decisions. For instance, the file export.coverage_stats.json has stats on the coverage of each term (1D-key) in the cohort. It is possible to go more granular with a tool like jq that parses JSON. For instance:
jq -r 'to_entries | map(.key + ": " + (.value | length | tostring))[]' < export.ref_hash.json
This command will print how many variables per individual were actually used to perform the comparison. You can post-process the output to check for unbalanced data.
Included R scripts
You can find in the link below a few examples to perform clustering and multidimensional scaling with your data:
Clustering
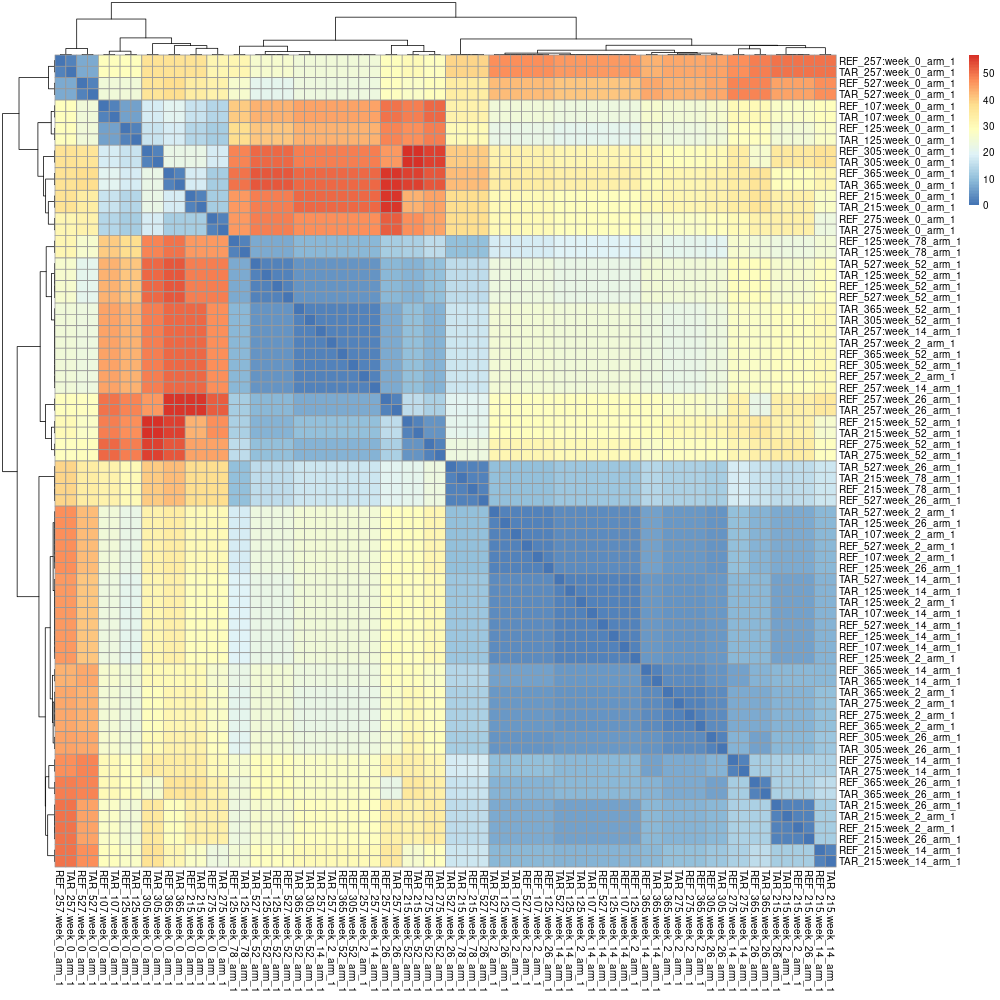
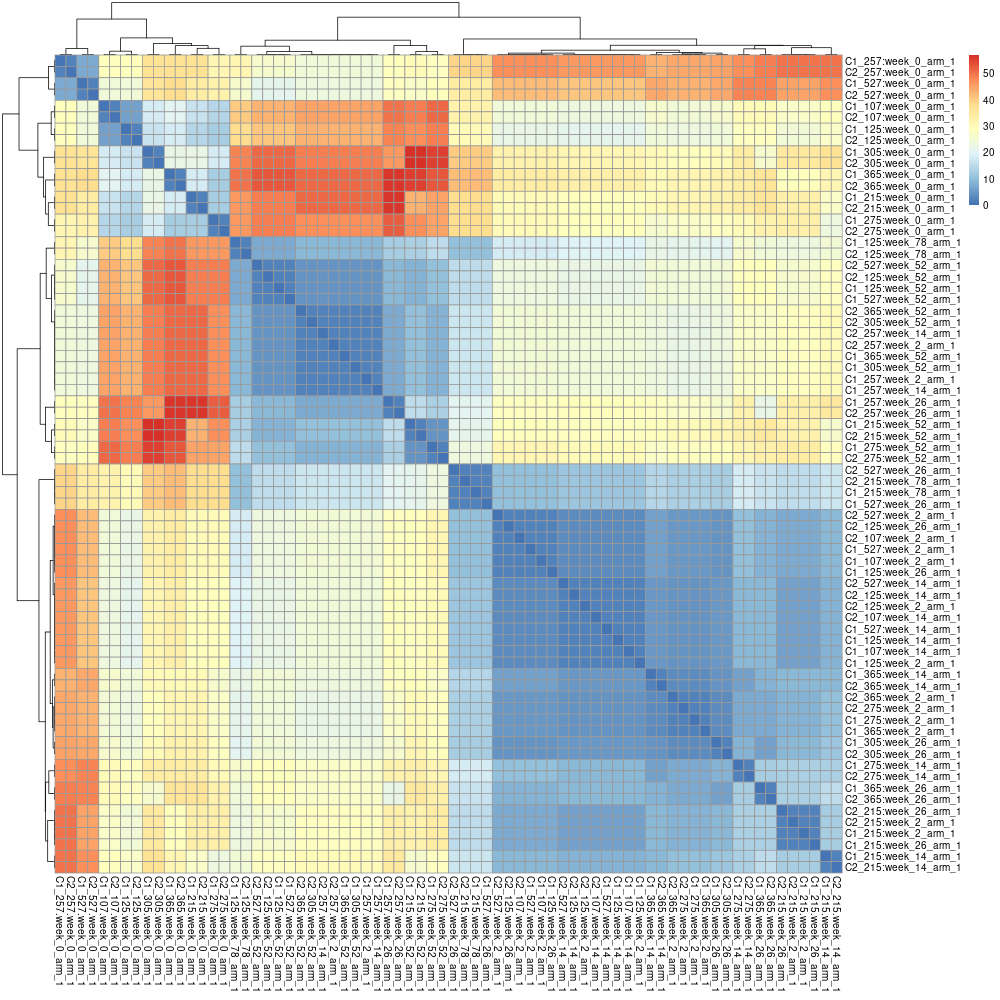
The matrix can be processed to obtain a heatmap:
R code
# Load library
library("pheatmap")
#library("heatmaply") # could not install
# Read in the input file as a matrix
data <- as.matrix(read.table("matrix.txt", header = TRUE, row.names = 1, check.names = FALSE))
# Save image
png(filename = "heatmap.png", width = 1000, height = 1000,
units = "px", pointsize = 12, bg = "white", res = NA)
# Create the heatmap with row and column labels
#heatmap(data, Rowv = FALSE, Colv = FALSE, labRow = rownames(data), labCol = colnames(data))
pheatmap(data)
#heatmaply(data)
#dev.off()
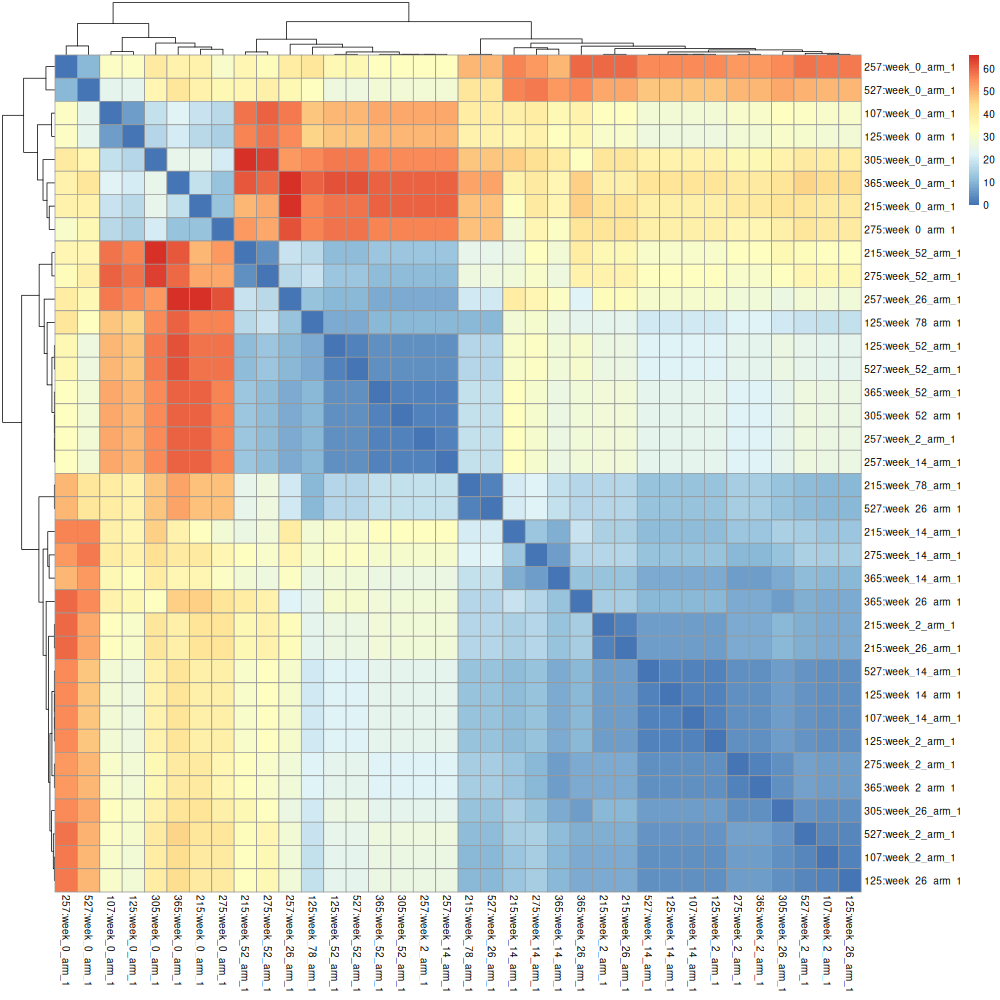
Dimensionality reduction
The same matrix can be processed with multidimensional scaling to reduce the dimensionality.
R code
library(ggplot2)
library(ggrepel)
# Read in the input file as a matrix
data <- as.matrix(read.table("matrix.txt", header = TRUE, row.names = 1, check.names = FALSE))
# Calculate distance matrix
#d <- dist(data)
#d <- 1 - data # J-similarity to J-distance
# Perform multidimensional scaling
#fit <- cmdscale(d, eig=TRUE, k=2)
fit <- cmdscale(data, eig=TRUE, k=2)
# Extract (x, y) coordinates of multidimensional scaling
x <- fit$points[,1]
y <- fit$points[,2]
# Create data frame
df <- data.frame(x, y, label=row.names(data))
# Save image
png(filename = "mds.png", width = 1000, height = 1000,
units = "px", pointsize = 12, bg = "white", res = NA)
# Create scatter plot
ggplot(df, aes(x, y, label = label)) +
geom_point() +
geom_text_repel(size = 5, # Adjust the size of the text
box.padding = 0.2, # Adjust the padding around the text
max.overlaps = 10) + # Change the maximum number of overlaps
labs(title = "Multidimensional Scaling Results",
x = "Hamming Distance MDS Coordinate 1",
y = "Hamming Distance MDS Coordinate 2") + # Add title and axis labels
theme(
plot.title = element_text(size = 30, face = "bold", hjust = 0.5),
axis.title = element_text(size = 25),
axis.text = element_text(size = 15))
#dev.off()
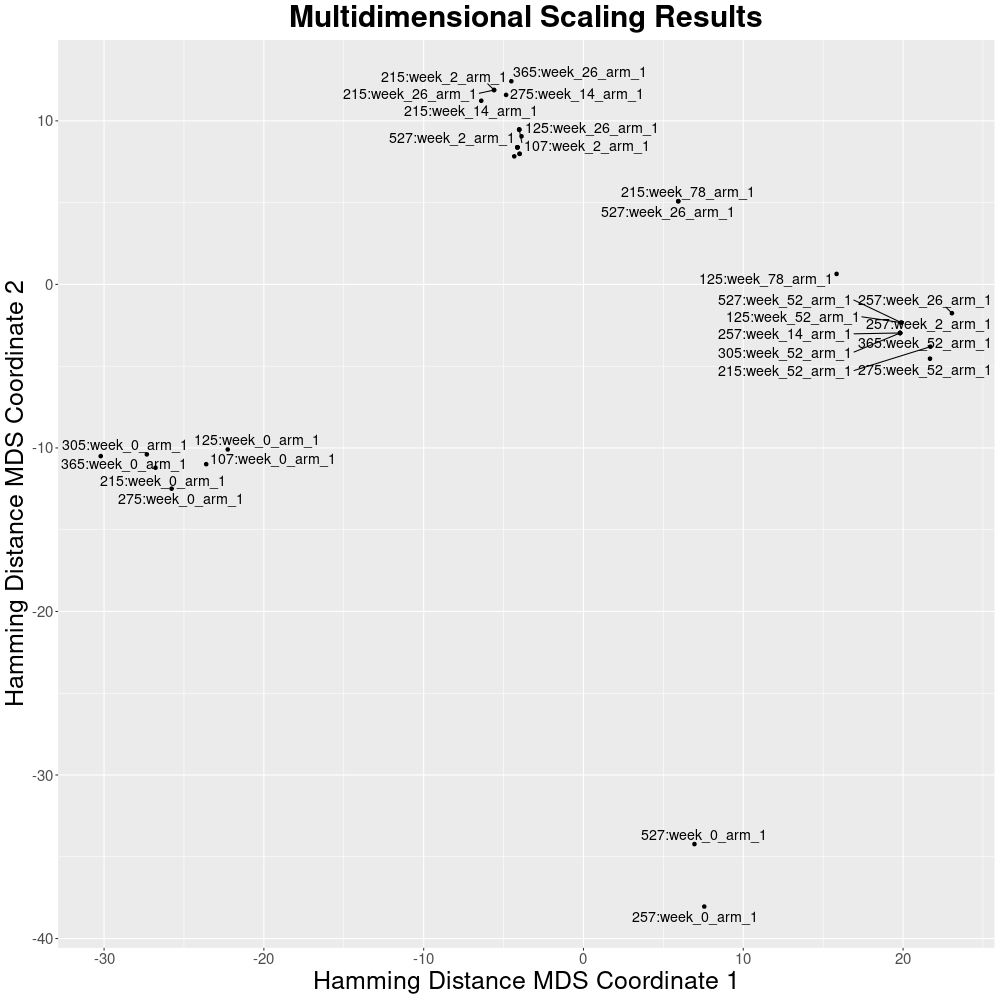
Or the dimensionality can be reduced with UMAP:
R code
# -- Install uwot on the fly if needed
if (!requireNamespace("uwot", quietly = TRUE)) {
install.packages("uwot", repos="https://cloud.r-project.org")
}
# -- Load libraries
library(uwot)
library(ggplot2)
library(ggrepel)
# -- Read in the input file as a full distance matrix
data <- as.matrix(
read.table("matrix.txt",
header=TRUE,
row.names=1,
check.names=FALSE)
)
# -- Convert to a 'dist' object so uwot knows these are distances
d <- as.dist(data)
# -- Set seed for reproducibility
set.seed(42)
# -- Run UMAP directly on the distances
# Passing a 'dist' object lets uwot build the k-NN graph from your distances
umap_res <- umap(
d,
n_neighbors=30,
min_dist=0.3,
n_components=2
)
# -- Extract UMAP coordinates
x <- umap_res[,1]
y <- umap_res[,2]
# -- Build a data frame for plotting
df <- data.frame(
x=x,
y=y,
label=rownames(data)
)
# -- Open PNG device
png(filename="umap.png",
width=1000,
height=1000,
units="px",
pointsize=12,
bg="white",
res=NA)
# -- Create scatter plot with labels
ggplot(df, aes(x=x, y=y, label=label)) +
geom_point() +
geom_text_repel(
size=5,
box.padding=0.2,
max.overlaps=10
) +
labs(
title="UMAP Embedding of Hamming Distance Matrix",
x="UMAP Coordinate 1",
y="UMAP Coordinate 2"
) +
theme(
plot.title=element_text(size=30, face="bold", hjust=0.5),
axis.title=element_text(size=25),
axis.text=element_text(size=15)
)
# -- Close the device
dev.off()
Graph analytics
Pheno-Ranker has an option for creating a graph in JSON format, compatible with the Cytoscape ecosystem.
Bash code for Cytoscape-compatible graph/network
pheno-ranker -r individuals.json --cytoscape-json
This command generates a graph.json file, as well as a matrix.txt file. The graph is generated directly from the binary comparison hashes, not by parsing the matrix file, so it can also be combined with Matrix Market output:
pheno-ranker -r individuals.json --matrix-format mtx -o matrix.mtx --cytoscape-json graph.json
Large graphs can be filtered by edge weight:
# Hamming distance: keep close pairs
pheno-ranker -r individuals.json --cytoscape-json --graph-max-weight 10
# Jaccard similarity: keep highly similar pairs
pheno-ranker -r individuals.json --similarity-metric-cohort jaccard --cytoscape-json --graph-min-weight 0.7
To produce summary statistics, use:
pheno-ranker -r individuals.json --cytoscape-json --graph-stats
This command will produce a file called graph_stats.txt. For additional information, see the generic JSON tutorial.
We use individuals.json again, but pass it twice to simulate two cohorts. Pheno-Ranker adds a CX_ prefix to the primary_key values so each record can be traced back to its source cohort.
pheno-ranker -r individuals.json individuals.json
Absolutely, a cohort can indeed be composed of a single individual. This allows for an analysis involving both a cohort and specific patient(s) simultaneously.
The prefixes can be changed with the flag --append-prefixes:
pheno-ranker -r individuals.json individuals.json --append-prefixes REF TAR
This will create a matrix.txt file of (36+36) x (36+36) cells. Again, this matrix can be processed with R: